
Annals of Medical & Surgical Case Reports
Review Article
Genetic and Hereditary Disorders in Iraqi Children
Al-Mosawi AJ *
Children Teaching Hospital, Baghdad Medical City, Baghdad, Iraq
*Corresponding author: Aamir Jalal Al Mosawi, Children Teaching Hospital, Baghdad Medical City, Baghdad, Iraq, Tel: +964770393083; Email: almosawiaj@yahoo.com
Citation: Al-Mosawi AJ (2019) Genetic and Hereditary Disorders in Iraqi Children. Ann Med & Surg Case Rep: AMSCR-1000011.
Received date: 06 August 2019; Accepted date: 15 August, 2019; Published date: 23 August 2019
Abstract
Background: Little is known about the uncommon, rare, and very rare genetic and hereditary disorders in Iraqi children. The aim of this paper is to describe the pattern of uncommon, rare and very rare genetic and hereditary disorders and hereditary disorders observed by one pediatrician at single tertiary pediatric center during three-year period.
Materials and Methods: During three-year period (2016-2018), 43 patients (29 males, 14 females) with uncommon, rare and very rare genetic and hereditary disorders were observed by one pediatrician at one tertiary pediatric center. Their ages ranged from 5 days to 17 years.
Results: Five patients had Duchenne muscular dystrophy, four patients had Charcot Marie Tooth disease, three patients had Wohlfart Kugelberg Welander, two brothers had Wiskott Aldrich syndrome, two sisters had hereditary (Familial) spastic paraplegia, two patients had Beckwith Wiedemann syndrome.
Fifteen unrelated male’s patients each had Berry-Treacher Collins syndrome, Pallister Hall syndrome, Ekman-Lobstein syndrome, Morquio syndrome, Werding Hoffman disease, Prader-Labhart-Willi syndrome, hypohidrotic ectodermal dysplasia, Lesch Nyhan syndrome, Pediatric Huntington disease, Sanjad Sakati Richardson Kirk Syndrome, Von Recklinghausen syndrome, Job’s syndrome, Autosomal recessive autism, phenylketonuria, homocystinuria.
Ten unrelated female patients each had cystinuria, Bequez Cesar syndrome, Maroteaux Lamy syndrome, Hurler Scheie syndrome, Adams Oliver syndrome, Townes Brocks syndrome, Classical pediatric Bartter syndrome, Coffin Siris syndrome, Cutis laxa type II (Debre type), Rett syndrome
Conclusion: In this series, very rare genetic and hereditary disorders in Iraqi children included the thirtysix case of Cutis laxa type II (Debre type) in the world, the case number 104 in the world of Sanjad-Sakati-Richardson-Kirk syndrome, the case number 130 in the world of Townes Brocks syndrome, and the case number 170 in the world of Coffin Siris syndrome.
Keywords: Children Rare; Genetic and hereditary disorders; Iraq
Introduction
Common genetic and hereditary disorders in Iraq include the thalassaemias and hemophilias [1,2], for these disorders specialized clinics and centers have been established. In addition to Down syndrome which has been observed frequently [2]. many other less common disorders have also been observed including achondroplasia, polycystic kidney disease, Duchenne muscular dystrophy, Werding Hoffman disease, Gaucher disease, mucopolysaccharidosis [1,3].
The occurrence of oculo-cerebro-renal syndrome, and nephropathic cystinosis have been well documented and studied in Iraqi children [4,5]. In fact, other genetic renal diseases (Table 1) have also been studied and reported [6-11].
Rare and very rare previously well documented genetic disorders in Iraq included diastrophic dysplasia, Coffin Siris syndrome, Cutis laxa type II (Debre type), Aicardi syndrome, and diastrophic dysplasia [12-17]. Little is known about the uncommon, rare, and very rare genetic and hereditary disorders in Iraq. The aim of this paper is to study the pattern of rare and very rare genetic disorders observed by one pediatrician at a single tertiary pediatric center during three-year period.
Materials and Methods
During three-year period (2016-2018), 43 patients (29 males, 14 females) with uncommon, rare and very rare genetic and hereditary disorders were observed by one pediatrician at one tertiary pediatric center. Their ages ranged from 5 days to 17 years.
Results
Five patients including brothers from three unrelated families had Duchenne muscular dystrophy. One of the patients had also autism, and one patient had bilateral testicular atrophy. Four patients including a brother and sister from two unrelated families had Charcot Marie Tooth disease.
Three unrelated male patients had Wohlfart Kugelberg Welander (Juvenile spinal muscular atrophy). Two brothers had Wiskott Aldrich syndrome. Two sisters had hereditary (Familial) spastic paraplegia. One of the sisters developed systemic lupus erythematosus. Two unrelated male patients had Beckwith Wiedemann syndrome (Figure 1A,B). Fourteen unrelated male’s patients had fifteen different disorders and ten unrelated female patients had different disorders (Table 2).
Discussion
There are few thousands of rare diseases affecting humans. Coffin Siris syndrome is a rare genetic disorder associated with mental retardation, coarse facial features, thick eyebrows, big mouth, hirsutism, and sparse scalp hair especially in the temporal region despite hypertrichosis [13,14]. Cutis laxa type II is a rare congenital disorder first described by Robert Debre, a French physician and his colleagues in 1937. is characterized by the unique association of generalized cutis laxa (loose skin from birth with redundant folds, generally sparing the face), characteristic facial features, late closure of the fontanel, and dislocation of one hip, pre-and postnatal growth retardation, and delayed motor development [15,16]. Aicardi syndrome is a rare genetic disorder of cerebro-ocular malformations that affects primarily females. It was thought that the syndrome is the result of heterozygous mutations in an X-linked gene, therefore it occurs only in persons with two X chromosomes. The disorder was probably first described by Jean Aicardi and colleagues in 1965. The syndrome is associated with agenesis of the corpus callosum, mental retardation, seizures and infantile spasms, and ocular abnormalities including chorioretinal lacunae, and microphthalmia [18]. Sanjad-Sakati-Richardson-Kirk is a syndrome of severe growth retardation, mental retardation, and chronic hypocalcemia caused by hypoparathyroidism was first reported by Sanjad, Sakati, and Abu-Osba in 1988, but it was first fully described in 1990 by Ricky J Richardson from the Sick Children Hospital of Great Ormond Street in London, and Jeremy MW Kirk from St Bartholomew's Hospital in London [19]. Townes Brocks syndrome is a rare autosomal dominant hereditary disorder which was probably first described in 1972 by Dr Philip L. Townes and Dr Eric Brocks. Dr Philip was professor of pediatrics at the University of Rochester, and Eric Brocks was a medical student. The syndrome is characterized by a triad of imperforate anus, limb defects, and ear abnormalities [20].
Very rare previously well documented genetic disorders in Iraq included the first case of Coffin Siris syndrome in the Arab which was the 76th or 80th case in the world depending of the exclusion of some doubtful cases [13,14,21], the Thirty fifth case of Cutis laxa type II (Debre type) in the world [15,22], the first case of Aicardi syndrome in an Arab patient which was associated with novel occurrence of corneal opacity [16].
In this series, very rare genetic disorders in Iraq included four very rare disorder. The Thirty-six case of cutis laxa type II (Debre type) in the world, and the second case in Iraq. The case was also most probably the first case to be associated with atrial septal defect [22]. The first case of Sanjad-Sakati-Richardson-Kirk syndrome in Iraq which was the case number 104 in the world [23]. The first case of Townes Brocks syndrome in Iraq which was the case number 130 in the world [20]. The second case of Coffin Siris syndrome in Iraq which was also the case number 170 in the world [21].
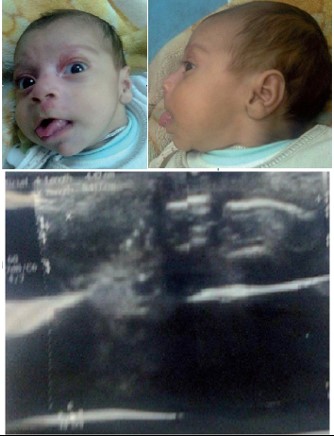
Figure 1A: The first patient with Beckwith Wiedemann syndrome. He had omphalocele at birth, recurrent hypoglycemia during infancy, macroglossia, and nevus flammeus. Abdominal ultrasound before the operation showed evidence of abdominal wall defect at the center of the abdomen measuring 4.4 cm, reducible and containing bowel loops.
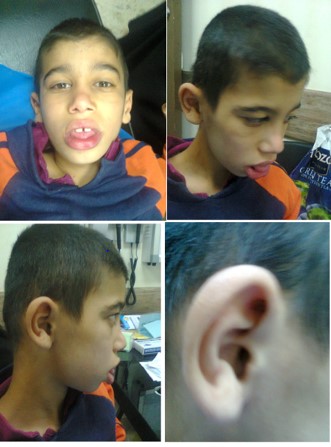
Figure 1B: The second patient with Beckwith Wiedemann syndrome. He had mental retardation, macroglossia, hepatomegaly, and recurrent hypoglycemia.
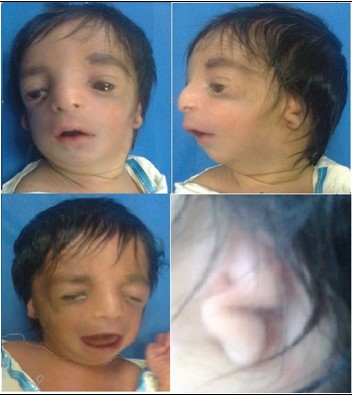
Figure 2: The patient with Berry-Treacher Collins syndrome had characteristic facial features including downward and laterally slanting palpebral fissures, paucity of lashes and lack of naso-frontal angle, bird like appearance, micrognathia, microtia (Deformed pinna), macrostomia and large tongue.
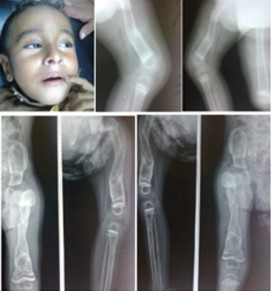
Figure 3: A boy with Ekman-Lobstein syndrome who had blue sclera and multiple recurrent fractures since early infancy. Radiographs showed osteoporotic changes, bowing of bones and multiple fractures.
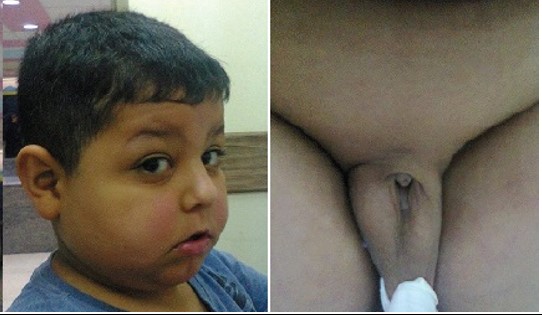
Figure 4: The patient with Prader-Labhardt-Willi syndrome. He mental retardation, almond eyes and a fish shaped mouth, and hypogonadism with extremely small penis that is hardly visible.
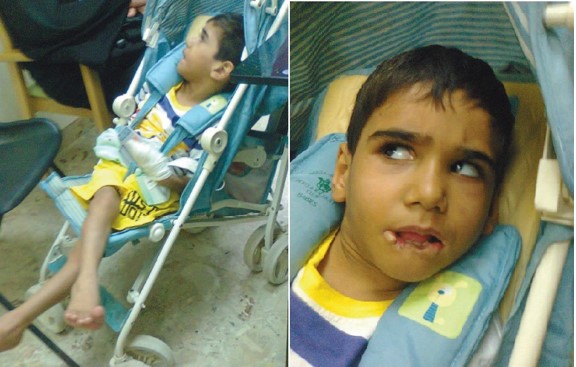
Figure 5: A nine-year old boy with Lesch Nyhan syndrome. His older brother died at the age of 14 years from the same condition. He had spasticity with scissoring of the lower limbs, choreoathetosis, self-mutilation with biting of the lips and hands, hyperuricemia, and renal stones.
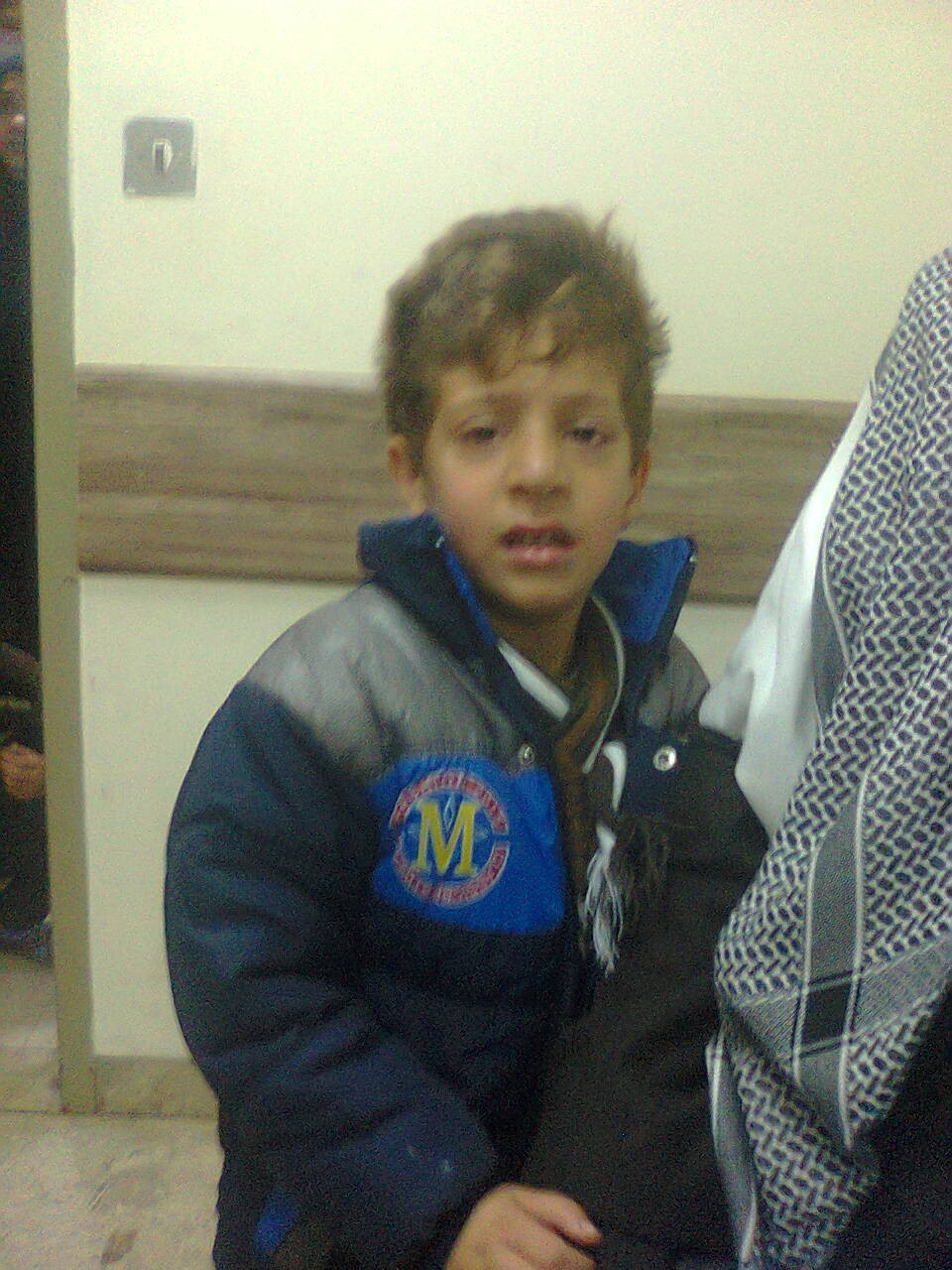
Figure 6: A boy with phenylketonuria and delayed institution of dietary restriction. He had mental retardation, light hair, hyperactivity, and history of seizures.

Figure 7: A ten-year boy with homocystinuria. He had mental retardation, posterior dislocation of the lens of the right eye, and markedly elevated serum methionine. He couldn’t copy a straight line.
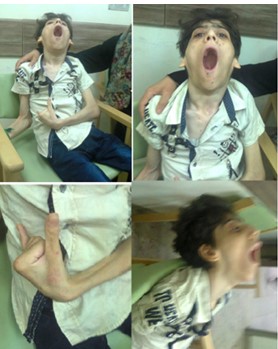
Figure 8: A 17-year boy with pediatric Huntington. He was unable to walk, unable to eat, unable to say a single word, and his mouth was kept open most of the time. He had choreoathetoid movements, and was developing recurrent extensor spasm of the neck.

Figure 9: An eight-year old boy with Sanjad-Sakati-Richardson-Kirk. He had chronic hypocalcemia, micrognathia, deep set eyes, thin lips, long philtrum, and beaked nose.
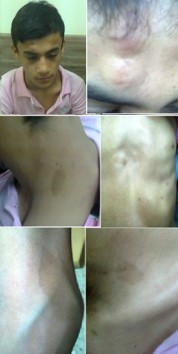
Figure 10: The boy with von Recklinghausen syndrome. He had soft tissue mass on the right forehead (neurofibroma). He also had multiple café au lait spots over many parts of his body including the back of neck, the anterior chest wall, back, arm and supra-clavicular region.
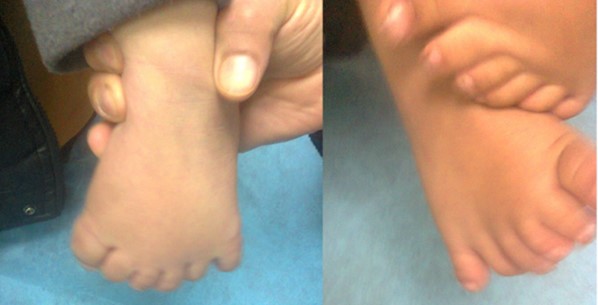
Figure 11: A boy with Pallister Hall syndrome who had imperforate anus at birth and polydactyly of hands and feet.
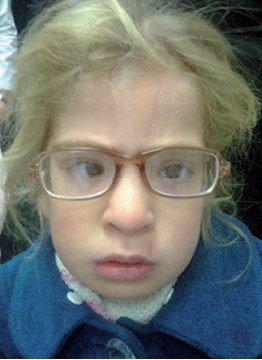
Figure 12: A girl with Beguez Cesar syndrome. She had recurrent infections, delayed development with poor speech, fair light skin, blond hair, photophobia, nystagmus, and a refractive error.
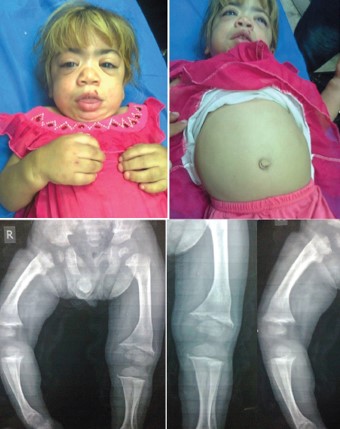
Figure 13: A girl with Maroteaux Lamy syndrome. She had coarse facial features including thick eyebrows, flat nasal bridge, thick lips, large mouth and tongue. She also had hepatosplenomegaly without mental retardation. She presented with difficulty in walking because of stiff joints and contractures at the knees, and radiographs showed evidence of skeletal dysplasia with bone enlargement, irregular shape and bowing.
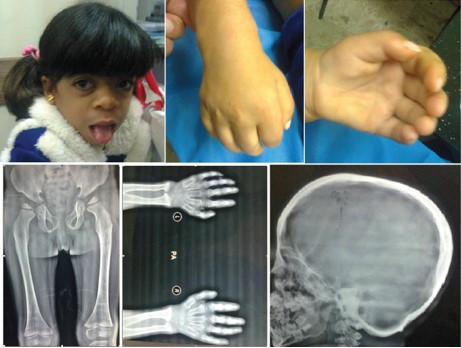
Figure 14: A girl with Hurler Scheie syndrome. She had coarse facial features including thick eyebrows, flat nasal bridge, thick lips, large mouth and tongue. She had difficulty in walking because of stiff joints and contractures at the knees, and radiographs showed evidence of skeletal dysplasia with bone enlargement, irregular shape and bowing she also had stiff joints with contractures at the hands. Radiograph also showed thickened calvarium. In addition, she had mitral and aortic regurgitation.
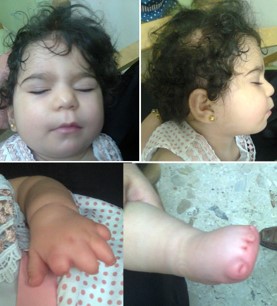
Figure 15: A girl with Adams Oliver syndrome. She had terminal transverse limb defects, hairless regions in the scalp and low set ears, she also had supra-valvular aortic stenosis which resulted in heart failure, and periventricular calcifications.
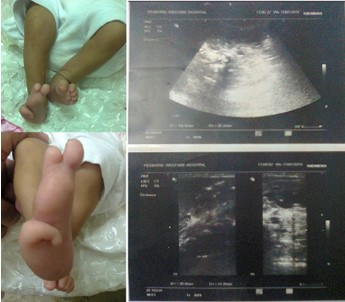
Figure 16: A girl with Townes Brocks syndrome. She had imperforated anus and needed colostomy. She also had deformity of the right foot with the presence of only three toes. Abdominal ultrasound small hypoplastic right kidney (18 × 12 mm) with normal shape.
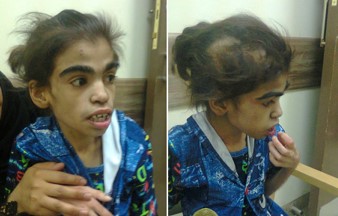
Figure 17: A twelve-year old girl with Coffin Siris syndrome. She had feeding difficulties, growth retardation, mental retardation and characteristic dysmorphic facial features including thick eyebrows, depressed and wide nasal bridge, low set ears, large mouth with thick everted upper and lower lips. Despite having hypertrichosis and hirsutism, she has area of hair loss.
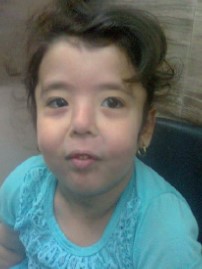
Figure 18: A girl with cutis laxa type II (Debre type). She had the characteristic redundant loose skin present since birth which was evident on the limbs and abdomen. She also had hypertelorism, depressed flat nasal bridge, high palate, and atrial septal defect.
|
Genetic renal diseases previously studied and reported in Iraqi children |
|
Oculo-cerebro-renal syndrome |
|
Nephropathic cystinosis |
|
Nephronophthisis |
|
Adult polycystic |
|
kidney disease |
|
Polycystic disease of kidneys and liver |
|
Primary oxalosis |
|
Cystinuria |
|
Autosomal recessive steroid resistant syndrome |
|
Autosomal recessive proximal renal tubular acidosis |
|
Autosomal recessive distal renal tubular acidosis |
|
X-linked dominant hypophosphatemic rickets |
|
Laurence-Moon-Biedl syndrome |
Table 1: Genetic renal diseases previously studied and reported in Iraqi children.
|
The number and sex of each genetic and hereditary disorders observed |
|||
|
Disorder |
Male |
Female |
Total |
|
Duchenne muscular dystrophy |
5 |
- |
5 |
|
Charcot Marie Tooth disease |
2 |
2 |
4 |
|
Wohlfart Kugelberg Welander |
3 |
- |
3 |
|
Wiskott Aldrich syndrome |
2 |
- |
2 |
|
Hereditary (Familial) spastic paraplegia |
- |
2 |
2 |
|
Berry-Treacher Collins syndrome (Figure 2) |
1 |
- |
1 |
|
Ekman-Lobstein syndrome (Figure 3) |
1 |
- |
1 |
|
Morquio syndrome |
1 |
- |
1 |
|
Werding Hoffman disease |
1 |
- |
1 |
|
Prader-Labhart-Willi syndrome (Figure 4) |
1 |
- |
1 |
|
Hypohidrotic ectodermal dysplasia |
1 |
- |
1 |
|
Lesch Nyhan syndrome (Figure 5) |
1 |
- |
1 |
|
Phenylketonuria (Figure 6) |
1 |
- |
1 |
|
Homocystinuria (Figure 7) |
1 |
- |
1 |
|
Pediatric Huntington disease (Figure 8) |
1 |
- |
1 |
|
Sanjad-Sakati-Richardson-Kirk Syndrome (Figure 9) |
1 |
- |
1 |
|
Von Recklinghausen syndrome (Figure 10) |
1 |
- |
1 |
|
Job’s syndrome |
1 |
- |
1 |
|
Autosomal recessive autism |
1 |
- |
1 |
|
Pallister Hall syndrome (Figure 11) |
1 |
- |
1 |
|
Cystinuria |
- |
1 |
1 |
|
Bequez Cesar syndrome (Figure 12) |
- |
1 |
1 |
|
Maroteaux Lamy syndrome (Figure 13) |
- |
1 |
1 |
|
Hurler Scheie syndrome (Figure 14) |
- |
1 |
1 |
|
Adams Oliver syndrome (Figure 15) |
- |
1 |
1 |
|
Townes Brocks syndrome (Figure 16) |
- |
1 |
1 |
|
Classical pediatric Bartter syndrome |
- |
1 |
1 |
|
Coffin Siris syndrome (Figure 17) |
- |
1 |
1 |
|
Cutis laxa type II (Debre type) (Figure 18) |
- |
1 |
1 |
|
Rett syndrome |
- |
1 |
1 |
Table 2: Shows the number and sex of each genetic and hereditary disorders observed.
Citation: Al-Mosawi AJ (2019) Genetic and Hereditary Disorders in Iraqi Children. Ann Med & Surg Case Rep: AMSCR-1000011.
