
Current Trends in Genetics and Microbiology
Complete Genome Sequence of Agrobacterium sp. ATCC 31749 and Insights into its Curdlan Biosynthesis
Anane RF1#, Lin C1,2#, Sun H1, Zhao L1, Liu Z1 and Mao Z1,2*
#These authors equally contributed to the study
1Department of Biochemistry, Physiology and Biotechnology, Yunnan Agricultural University, Kunming, China
2Institute of Improvement and Utilization of Characteristic Resource Plants, Yunnan Agricultural University, Kunming, China
*Corresponding author: Mao Z, Department of Biochemistry, Physiology and Biotechnology, Yunnan Agricultural University, Panlong District, Kunming, 650201, China, Tel: +8613114297551.
Citation: Anane RF, Lin C, Sun H, Zhao L, Liu Z, et al. (2019) Complete Genome Sequence of Agrobacterium sp. ATCC 31749 and Insights into its Curdlan Biosynthesis. Curr Trends Genet Microbiol: CTGM-100001
Received date: 18 February, 2019; Accepted date: 5 March, 2019; Published date: 20 March, 2019
Abstract
Curdlan is an important glucan that is extensively used in food and pharmaceutical industries for production of food and medicines, and its natural producer is the Agrobacterium sp. ATCC 31749. Understanding its complete genome will provide great insights for the scientific community and industrial producers of curdlan. This study provides comprehensive knowledge of the genome architecture of Agrobacterium sp. ATCC 31749, highlighting the genetic basis for which Agrobacterium sp. ATCC 31749 effectively produces curdlan and the evolutionary relationships of Agrobacterium sp. ATCC 31749 with other species within the family Rhizobiaceae. Agrobacterium sp. ATCC 31749 produces curdlan as a protective glucan to protect itself against unfavourable conditions. The complete genome structure of Agrobacterium sp. ATCC 931749 consists of two small circular plasmids, one large primary chromosome and one secondary chromosome. Genes that are responsible for housekeeping activities and basic life processes are located on the primary chromosome, although curdlan biosynthesis genes are located on the secondary chromosome. Integration of fragments of an ancestral symbiosis/Ti plasmid into a duplicated copy of the ancestral primary chromosome led to formation of the secondary chromosome during the process of evolution. The secondary chromosome of Agrobacterium sp. ATCC 31749 contains a plasmid-type replication origin similar to that of a Ti plasmid. Genes necessary for regulatory mechanisms that improve expression of crucial genes that enhance curdlan biosynthesis in response to environmental stress stimuli are consistently upregulated with curdlan biosynthesis operon genes in the stationary growth phase. The SNPs changes observed in the curdlan biosynthesis operon are vital for curdlan biosynthesis in Agrobacterium sp. ATCC 31749.The closest Agrobacterium species to A. sp. ATCC 31749 is the pathogenic A. fabrum str. C58. This study offers further insight into the genetic mechanisms for curdlan biosynthesis and regulation, as well as the genetic basis for bioengineering of this species by commercial producers of curdlan for higher production of curdlan.
Keyword: Agrobacterium sp; ATCC 31749; Curdlan; Genome; Multichromosomal bacteria; Secondary chromosome; Origin of replication
Abbreviations: ATCC31749; C1; C2, LC34; SUL3; C58
Introduction
The capability of microorganisms to successfully adapt and cope with environmental constraints is dependent upon their successful production and secretion of both capsular and extracellular polysaccharides (CPS and EPS). EPS is crucial for bacteria pathogenicity, biofilm formation and survival during adverse environmental conditions [1-3] by serving as a protective boundary between cells and their immediate outer environment [4,5]. The presence of EPS on bacterial cells promotes bacterial longevity by extending the period for physiological adaptation to external changes. One of such important EPS with a wide range of applications in pharmaceutical and food industries is curdlan, a water-insoluble β-(1,3) glucan, which is commercially produced by Agrobacterium sp. ATCC 31749 (ATCC31749). ATCC31749 is an α-Proteobacteria that belongs to the family Rhizobiaceae in the order Rhizobiales, in which most organisms are either symbiotic nitrogen-fixing or pathogenic to their host plants [6-9]. Although ATCC31749 is an Agrobacterium possessing some ancestral symbiotic and invasion-regulatory-related genes, it is neither plant associated nor virulent to plants [10].
The establishment of a symbiotic relationship with a host plant demand additional genetic requirement obtained mainly through lateral gene transfer and/or diversification after gene duplication in order to adjust to the demands and challenges imposed by symbiotic/pathogenic relationship [11]. To by-pass the high genetic requirements for symbiosis and to simplify the genome requirements for life while maximizing its survival under stress conditions, ATCC31749 might have evolved curdlan biosynthesis genes to produce curdlan over the process of evolution and selection [12,13]. Curdlan does not only possess multi-functional protective ability, but also has the ability to envelope the ATCC31749 cells surfaces thereby providing means of adhesion to surfaces and protection against biotic and abiotic stresses - physical and chemical [14,15]. The high carbon content of curdlan makes it a good nutrient repository that provides carbon source and stability to the cell surface to offer protection for the cells against wilting. Understanding the complete genome structure, genetic basis and location of primary regulons in the genome of ATCC31749 will provide great insights into its metabolic engineering to improve curdlan biosynthesis and transportation from intracellular to extracellular.
Here, we report that the complete genome of ATCC31749 consist of two chromosomes and two plasmids. It is noteworthy to state that genes that are responsible for housekeeping activities and basic life processes are located on the primary chromosome, although curdlan biosynthesis genes are located on the secondary chromosome. We postulate that the secondary chromosome of ATCC31749 is derived from a duplicated copy of the primary chromosome to which fragments (including the origin of replication) of an ancestral symbiosis/Ti plasmid has integrated into. Furthermore, our results show that the ATCC31749 genome resembles more closely to the genomes of pathogenic Agrobacterium species than non-pathogenic Agrobacterium species and hence, our phylogenetic tree locates ATCC31749 to be grouped with pathogenic Agrobacterium species. Agrobacterium fabrum str. C58 is the closest related Agrobacterium species to ATCC31749 and they may have a common pathogenic/symbiotic Rhizobiales genetic ancestor. This study therefore provides comprehensive knowledge on the genome architecture of ATCC31749, highlighting the evolutionary relationships of ATCC31749 with other species within the family Rhizobiaceae. This study also provides insights into the genetic basis for which ATCC31749 effectively produces curdlan. The insights obtained from the secondary chromosome that bears the curdlan biosynthesis operon and the SNPs changes observed in this operon, as well as the replication origin (plasmid-based origin) of the secondary chromosome will be helpful for bioengineering this species. Furthermore, the evolutional changes of symbiosis-related genes, such as nodulation, nitrogen fixation, secretory systems, and EPS synthesis and transport related genes were thoroughly investigated to understand the evolution of ATCC31749. The complete genome sequence of Agrobacterium sp. ATCC 31749 has been deposited at NCBI with GenBank submission ID 2188667.
Methods
Chemicals, bacteria strains, culture and genomic DNA extraction
All chemicals used in this study were obtained from Sangon Biotech, China. The bacteria strain Agrobacterium sp. ATCC 31749 was obtained from the American Type Culture Collection (ATCC, Manassas, VA, USA). It was stored in a complex medium in −80°C refrigerator in Mao Zichao’s laboratory in the College of Agronomy and Biotechnology, Yunnan Agricultural University, Kunming, China. ATCC31749 cells were cultured at 30°C for 20 h on LB agar media. A single colony from this culture was inoculated into 5 ml LB broth and incubated in a shaker at 30°C, 200rpm for 24 h. After that a 1:50 dilution culture was performed in 100 ml fresh LB broth which was incubated in a 30°C shaker at 200rpm for 20 h. Extraction of genomic DNA was performed as described by Wilson [16]. Cells were harvested and lysed by EDTA, lysozyme, and detergent treatment, followed by proteinase K and RNase digestion. DNA isolation was done by phenol-chloroform-isoamyl alcohol extraction followed by repeated iso-propanol-ethanol precipitation. DNA quality was determined by agarose gel electrophoresis and the purity was measured at the A260/A280 ratio.
Genome sequencing, assembly and annotation
ATCC31749 genome was first sequenced by pair ends using the Illumina Hiseq 2500 sequencing platform with 500 bp and 3000 bp insertion library to obtained a random 217 X coverage data which was used for assembly by SOAPdenovo2 (http://soap.genomics.org.cn/soapdenovo.html) with default parameters. The resulting assembled scaffolds were compared with the scaffolds obtained by Ruffing (10) to improve the draft genome assemble of ATCC31749. Closing of gaps in the genome was performed by SMRT sequencing [17] to obtain 20 X data with 20 kb size insertion library, followed by genome assembly with SPAdes 3.11 software (http://bioinf.spbau.ru/spades/). RNAmmer [18], tRNAscan-SE [19], Prodigal software [20] and TRF (http://tandem.bu.edu/trf/trf.html) were used for gene prediction and genomic repeat elements analysis. Prior to manual functional annotation, an automatic annotation was computed based on different tools by BLAST search against the following databases: Non-redundant proteins database (NR) [21], Cluster of Orthologous Groups (COGs) [22], Gene Ontology (GO) and the Kyoto Encyclopedia of Genes and Genomes (KEGG) [23,24]. The complete genome structure and features were visualized with Circos software [25]. The CRISPRC as Finder online program (https://crisprcas.i2bc.paris-saclay.fr/CrisprCasFinder/Index) was used for detection of CRISPRs.
Phylogeny and comparative genome analysis
To analyze the phylogenetic position and molecular evolution of Agrobacterium sp. ATCC31749, the complete genome protein coding sequence of ATCC31749 was used to perform local BLASTP (at e-value of 1 x 10-7) against the complete genome protein coding sequences of other 56 members of the Agrobacterium/Rhizobium group by using BLASTP 2.7.1 software [26] to determine homologous proteins. The genome protein coding sequences of the other 56 species were downloaded from the NCBI website (ftp://ftp.ncbi.nlm.nih.gov/genomes/genbank/bacteria). After BLASTP analysis, the resulting data was filtered by two criteria (cut-off of 80% similarity and cut-off of 80% amino acid coverage) to select highly homologous proteins. The obtained protein sequences (encoded by 247 genes) of the 57 species were used for alignments and construction of phylogenetic tree to analyze the phylogenetic position of ATCC31749. Alignments and generation of phylogenetic trees from concatenated core gene alignments were performed with MAFFT version 7.4 at MAFFT online service (https://mafft.cbrc.jp/alignment/server/) [27] by using the Neighbor-Joining algorithm method and JTT model. The generated trees were viewed with the Interactive Tree of Life (iTOL) online tool (http://itol.embl.de/) [28].
Comparative genome analysis to study the orthologous clusters of genes between the genomes of ATCC31749, Agrobacterium fabrumstr. C58 (C58), A. fabrumstr. J-07 (J-07) and A. tumefaciens str. LBA4404 (LBA4404), A. sp. LC34 (LC34) and A. sp. SUL3 (SUL3) was performed by using the OrthVenn online tool (http://www.bioinfogenome.net/OrthoVenn/). Further comparative genome analysis between the genomes of ATCC31749 and C58 was performed by using the circoletto web server (http://bat.ina.certh.gr/tools/circoletto/) to construct syntenic relationship circos plot at e-value of 1x10-5. The plot was colored by the ‘score/max’ ratio of tBLASTxbitscores (real score/maximal score) with ribbon colors of blue ≤ 0.25, green ≤ 0.50, orange ≤ 0.75 and red > 0.75. To analyze the evolution of the secondary chromosome of ATCC31749, two phylogenetic trees inferred from OriC DNA nucleotide sequences of the primary and secondary chromosomes of 41 multichromosomal bacteria species were constructed.Secondly, a phylogenetic tree inferred from nucleotide sequences of the repABC genes of the replicons of ATCC31749, the secondary chromosome and plasmids of C58 (C2, pTiC58, pAtC58), plasmids of J-07 (pTiJ07, pAtJ07),LBA4404 (pTiLBA4404, pAtLBA4404), Rhizobium leguminosarum bv. TrifoliCB782 (pRtCB782a, pRtCB782b, pRtCB782c) and A. vitisS4 (pAtS4a, pAtS4b, pAtS4c, pAtS4e and pTiS4) was constructed. Finally, syntenic genes relationships plot between the chromosomes and plasmids of ATCC31749 was constructed by using the circoletto web server at e-value of 1x10-5. The plot was colored by the ‘score/max’ ratio of tBLASTxbitscores (real score/maximal score) with ribbon colors of blue ≤ 0.25, green ≤ 0.50, orange ≤ 0.75 and red > 0.75.
Analysis of differentially expressed genes of ATCC31749
A single colony of ATCC31749 from an overnight culture was inoculated into 5 mL LB broth medium and incubated overnight in a shaker at 30°C, 200 rpm. The resulting culture was used to inoculate a fresh 100 ml LB broth media in 500 ml flask with a dilution ratio of 1:50. The flask cultures were incubated at 30°C in a shaker (200 rpm) to a final optical density (OD600) of approximately 0.5, corresponding to the exponential growth phase (E-phase) cells. 20 ml replicates of these cell cultures were pelleted from the medium by centrifugation at 4000 × g for 5 min. The cell pellets were re-suspended in 100 mL curdlan fermentation media as previously described (15) for 24 h, which corresponds to the stationary growth phase (S-phase) cells. The rRNA-depleted RNA was prepared from both E-phase and S-phase cell pellets by the method developed by Chen and Duan [29]. The resulting RNA samples were sent to Biomarker (Beijing, China) Illumina Hiseq 2500 sequencing platform for RNA-sequencing. The obtained RNA-seq data was used for differential expression analysis by using HISAT2, StringTie and Ballgown RNA-seq analysis pipeline [30,31]. Analysis and generation of heat map for expression profiles of curdlan biosynthesis genes and their related regulatory genes at both E- and S-phases were performed by d3heatmap package of R.
Protective ability of curdlan on ATCC31749 cells against stress conditions
The cells of 3 different strains of ATCC31749 at both E-phase and S-phase: ATCC31749 (Curdlan-producing ATCC31749, wild type), ATCC31749?crdR (crdR knockout mutant which produces lower curdlan yield) and ATCC31749?crdR/pBQcrdR (pBQcrdR transformed strain for functional complementarity of crdR) were used to study the protective ability of curdlan on ATCC31749 cells against some stress conditions. The stress conditions investigated include high temperature (42?C), UV light and acidic condition. For investigation of high temperature, cells of the 3 strains that have been grown to both E- and S-stages were spread unto LB agar plates and placed in a 42?C incubator for 48 h. The cells were then placed in a 28?C incubator for another 48 h and the survived colonies were counted. For investigation of UV light, cells of the 3 strains that have been grown to both E- and S-stages were spread unto LB agar plates and placed under UV light for 30 minutes, after which the plates were incubated at 28?C for 48 h and the colonies that appeared were counted. To investigate the protective ability of curdlan on ATCC31749 cells in acidic condition (pH=2-3), cells of the 3 strains that have been grown to both E- and S-phases were spread unto LB agar plates (pH=2-3) and incubated at 28?C for 48 h. The colonies that appeared were counted. A graph of number of colonies against stress condition at each growth phase was plotted.
Results
Genome sequencing, assembly and general features
The genome was sequenced using the Illumina Hiseq 2500 platform, generating a total 3.1 Gb clean data with an average qualified read size of 125 bp. The sequenced data (about 217 x coverage) was assembled by SOAPdenovo2 and compared with the draft genome contributed by Ruffing (10), resulting in a new draft genome with 14 scaffolds. The SPAdes software [32] was used to assemble the 20 x third generation data produced by Pacific Bioscience SMRT method with 20 kD insertion sizes library. Compared with the 14 scaffolds draft genome, the final complete genomic sequence was obtained. The complete genome has a GC content of 58.9%, and is composed of four DNA replicons of which two are chromosomes (one primary chromosome- the largest chromosome, C1; and one secondary chromosome, C2) and the other two are extra-chromosomal plasmids (pAg31749a and pAg31749b) (Table 1, Figure 1 and File S1). Out of the predicted 5591 protein-coding genes 4,441 (79.4%) are known functional genes whiles 1,150 (20.6%) are hypothetical genes. Details of the general features of the genome can be found in Table 1.According to the results of the COG annotation, the genome is enriched with genes that function in transport and metabolism of amino acid (E) and carbohydrate (G), and also in transcription (K) (Figure 2a). It is evident from the GO analysis that in cellular component function, most genes are either membrane, membrane part or cell part. In molecular function, most genes function in catalytic, binding and transport activities, whiles in biological process, genes mostly function in single organism processes, metabolic processes, cellular processes, response to stimulus, localization and biological regulations (Figure 2b). Since synthesis of curdlan by ATCC31749 is initiated by and effective under stress conditions, and that the raw materials (such as sucrose) used for synthesis of curdlan has to be transported to the intracellular, whiles the produced curdlan is transported to the extracellular environment; it is not surprising that the genome is rich in genes that encode proteins related to membrane transport and signaling components.
Evolution of Agrobacterium sp. ATCC31749
To investigate the evolutionary relationships between ATCC31749 and other members of the Agrobacterium/Rhizobium group, a phylogenetic tree inferred from protein sequences of the 247 highly conserved orthologous genes (File S2) was constructed with Neighbor-Joining algorithm method. 86% of the 247 genes were located on the primary chromosome whiles the other genes were located on the secondary chromosome. Analysis of the concatenated data set produces a single topology with a 100% posteriori support for all branches within the constructed Agrobacterium/Rhizobium group tree. The constructed phylogenetic tree finds ATCC31749 to be placed in the clade (C2) containing three Agrobacterium biovar I species: C58, LBA4404 and J-07 (Figure 3). The Neighbor-Joining algorithm tree gives the same topology and similar relative branch lengths as the tree produced by Maximum likelihood analysis (Figure S1). It can be inferred from the phylogenetic tree that C58 is the closest related species to ATCC31749 (Figure 3 and Figure S1), although C58 is pathogenic (contains a Tiplasmid) whereas ATCC31749 is not. Comparative genome analysis for orthologous clusters of genes between the genomes of the species of “clade B2” in figure 3 shows that, 3698 orthologous clusters of genes are shared between the six species. 3708, 3701, 3807, 3704 and 3724 orthologous clusters are shared between ATCC31749and C58, ATCC31749 and LBA4404, ATCC31749 and J-07, ATCC31749 and LC34, ATCC31749 and SUL3 respectively (Figure 4a). This result explains that ATCC31749 genome shares more homologous clusters with pathogenic species (clade C2) and therefore resembles more closely to the genomes of pathogenic Agrobacterium species than that of the nonpathogenic species (clade C1) of Figure 3.
Analysis of syntenic genes relationships between replicons of ATCC31749 and C58 reveals that ATCC31749_C1 shares very high homology to C58_C1, whiles ATCC31749_C2 is more homologous to C58_C2 with some genes being mapped to pTiC58. Similarly, the plasmid pAg31749a is highly mapped to the plasmid pAtC58. Surprisingly, plasmid pAg31749b is highly mapped to C58_C2 with very few fragments being mapped to pAtC58 and pTiC58 (Figure 4b). Analysis of dnaA (chromosomal replication initiator gene) and oriC (chromosomal origin of replication gene) which are necessary for initiation of chromosomal replication in bacteria reveals that C1_oriC of the primary chromosome of ATCC31749 has a 92.252% (e-value < 1 x 10-7) identity to the known oriC of C58, whiles the C2_oriC of the secondary chromosome has 97.208% identity to that of C58. The dnaA protein sequence of ATCC31749 and C58 share 100% identity. It is therefore possibility that ATCC31749 may be a sub-species of the Agrobacterium fabrum group and that ATCC31749 may have a pathogenic genetic ancestor.
Comparative genomics
Since our molecular evolution tree reveals that C58 is a closely related species to ATCC31749 (Figure 3), some symbiotic/pathogenic related genes, stress responsive genes, and CPS/EPS biosynthesis and transport related genes of C58 were searched in the genome of ATCC31749. Some homologous of symbiotic/pathogenic-related genes of C58 such as invasion-associated regulatory genes and damage-inducible genes including nodulation genes (nodN, nodT, nolG, nolR), nitrogen-fixation-regulatory related genes (fixL, fixG, fixH, nifR, nifH,NtrBC, NtrXY), virulence-associated and regulatory genes (PhoQ, PhoP, chvB,VirE, VirKfamily) and cytochrome oxidative gene were found to be present in ATCC31749 genome. Furthermore, stress responsive/tolerant and multi-chemical resistant genes such as AcrB/AcrD/AcrF families, the universal stress tolerance and defense protein-coding genes, such as Type I restriction modification genes, are also present in the ATCC31749 genome. Similarly, EPS and CPS biosynthesis related genes such as glycogen synthase, curdlan synthase operon (crdASC), exoAB, exoDF, exoHI, exoKLMN, exoOPQ, exoRST, exoUVZXYZ and chvEIG are located in theATCC31749 genome (File S3). The C58 genes chvAB (essential for synthesis and localization of the extracellular β-1,2-glucan which enable C58 cells to bind to plant cells) [33-35], ChvH (glucan elongation factor), ChvI (two component response regulator), chvE (sugar binding periplasmic gene), ChvG (two component sensor kinase) and exoR (exopolysaccharide synthesis repressor gene), which are all located on primary chromosome of C58 are also found to be anchored on the primary chromosome of ATCC31749. Similarly, the secondary chromosome of ATCC31749 harbors the cel (cellulose synthase gene) and exoC gene required for synthesis of extracellular β-1,2-glucan and succinoglucan polysaccharides. The β-1,2-glucan synthase genes of ATCC31749 and C58 share 99.74% identity. Polysaccharide synthesis related genes such as pssA (Phosphatidylserine synthase) and pssB (EPS production gene), and pssN (EPS export gene) are also found to be anchored on the primary and secondary chromosomes respectively in ATCC31749. The repABC genes involved in replication initiation, copy number control, and partition of molecules are found in the secondary chromosome and plasmids in ATCC31749. These results confirm our molecular evolution tree analysis which suggests that ATCC31749 may have a symbiotic/pathogenic ancestor similar to that of C58.
Some major homologous genes for the regulation of virulence factors were found to be present in ATCC31749 genome, although ATCC31749 is not virulent. Chromosomal virulence gene chvB, GDP-mannose 4,6-dehydratase (WbkC), Glycosyl transferase family (WbkA), Phosphoglucomutase (Pgm), ABC transporter ATPase (Wzt), Mannose-1-phosphate guanyltransferase (ManCoAg) and Mannose-6-phosphate isomerase (ManAoAg) are located on the chromosomes of ATCC31749. Furthermore, principal constituents of protective antigens of Brucella spp. such as ArsR family transcriptional regulator (AsnC), Molecular chaperones (DnaK, SurA, DnaJ), Cobyrinic acid a,c-diamide synthase (CobB), Glyceraldehyde-3-phosphate dehydrogenase (GapA), Membrane protein gene (OmpA), Superoxide dismutase gene (SodC), Invasion-associated protein and Bacterioferritin (Ferritin) genes which are all necessary for production of Brucella vaccines [36] are also located in the genome of both C58 and ATCC31749. These results also support our hypothesis that both ATCC31749 and C58 may have common ancestors.
DNA and amino acids sequences of the curdlan synthesis operon genes (crdASC) of ATCC31749 were compared to C58 by BLAST analysis to understand the reasons for which ATCC31749 effectively produces curdlan. The crdA of both C58 and ATCC31749 has 12 SNPs changes and 1 insertion at the C-terminal coding region of crdAof C58 leading to reading frame-shift. The crdS has 22 SNPs changes between C58 and ATCC31749 leading to 5 amino acid changes (L64P, F66L, A140G, A305V, T586K) in ATCC31749. There were 21 SNPs changes observed between the crdA of C58 and ATCC31749 that results into 7 amino acid changes (S8G, G95Q, Y127H, M321I, L249S, V393I, R417S) in ATCC31749. These differences and changes observed in the crdASC operon could be some of the reasons for which ATCC31749 effectively produces curdlan under nitrogen limited conditions.
Primary chromosome (C1)
In multichromosomal bacteria, primary metabolism regulons and housekeeping genes are located on the primary chromosomes. To determine which of the chromosomes of ATCC31749 is the primary chromosome (C1), vital and primary metabolism related genes involved in most essential processes and housekeeping jobs in the life of a bacterium were analysed. The Central Dogma genes and the DNA recombination, replication and repair genes such as recA (DNA recombinase and repair protein A), atpD (ATP synthase subunit beta), glnD (bifunctional uridylyltransferase), rpoB (DNA-directed RNA polymerase subunit beta) and eftU (elongation factor Tu) were all found to be located in circular chromosome. The same locations were also found in C58, J-07 and LBA4404 in the clade “C2” of the phylogenic tree in figure 3. Another vital regulon in the bacteria domain is the SOS response genes which are responsible for repairing DNA damage, stress-induced mutagenesis, and development. Analysis of the locations of the three core genes of the SOS response (lexA, recA, uvrA) reveals that, the lexA (SOS transcription regulator), recA (DNA recombinase) and uvrA (excinuclease subunit A) genes are located on the circular chromosome of ATCC31749. These genes were also found to be located on the primary chromosomes of C58, J-07 and LBA4404, and Vibrocholerae and Brucella melitensis 16M belonging to the Proteobacteria group. Similarly, the location of a specialized triad machinery components (XerC, XerD and FtsK) which resolves DNA dimers in bacteria cells was analysed. These genes were also found to be anchored on the circular chromosome of ATCC31749. Other genes necessary for basic life of a bacteria cell such as DNA replication and replication initiator genes (ParA, ParB, ParC, ParE, DnaA), cell division and segregation genes (FtsY, FtsZ, FtsA, FtsQ, FtsW, MraZ), DNA repair genes (RecO, RecN, RecF, RecR,MutL, MutT, MutS), nucleotide and amino acid synthesis pathway and regulatory genes, DNA gyrase (gyrA, gyrB) and its inhibitor (YacG), ATPases (AAA) genes, and glycolysis related genes are mainly located on the circular chromosome of ATCC31749(File S3). Similarly, glutamine synthase (glnA, glnII), threonine synthase (thrc), transcription terminator/antiterminator (nusA), glucose-6-phosphate 1-dehydrogenase (zwf), anthranilate synthase (trpE), 60kDa chaperon in (groEL), and molecular chaperone DnaK (dnaK) genes are all located on the circular chromosome of ATCC31749. Furthermore, the circular chromosome of ATCC31749 contains a putative origin of replication (oriC) that has 92.252% identity to the known oriC of C58 primary chromosome. These results confirm that the circular chromosome is the primary chromosome of ATCC31749 and also supports the notion that in multichromosomal bacteria, primary regulons and genes that carry out most of the housekeeping and cellular activities of a cell are located on the primary chromosome [37].
Secondary chromosome (C2)
To understand the evolution of the secondary chromosome of ATCC31749, whole DNA nucleotide sequences of the replicons of ATCC31749were used to perform syntenic genes relationship analysis. The syntenic relationship tBLASTx graphical comparison of the chromosomes (C1, C2) with the plasmids indicate that, the secondary chromosome shares high level of similarity to the primary chromosome (Figure 5a), suggesting a possibility that C2 could have originated from duplication of the C1. The two phylogenetic trees constructed from the replication origin (OriC) DNA nucleotide sequences of the primary and secondary chromosomes (C1_oriC and C2_oriC) of 41 multichromosomal bacteria species of the Proteobacteria suggest that, the C1_oriC and C2_oriC of ATCC31749 are more homologous to their counterparts of C58. However, C1_oriC and C2_oriC of ATCC31749 are located at different clades that are very distant from each other (Figure S2a & S2b). This suggests that C1_oriC and C2_oriC of ATCC31749 have different origins of evolution, and that the origin of the secondary chromosome may have come from a remote ancestral replicon. Furthermore, comparative analysis of the C1_oriC, C2_oriC, pAg31749a_oriC and pAg31749b_oriC shows very low similarities between them, indicating that the replication origins of the plasmids and chromosomes of ATCC31749 may have evolved from different ancestors. Analysis of the C2_oriCof ATCC31749 reveals that the C2 has a plasmid-type replication system (repABC) which is located at the centre of the chromosome. The result of BLASTN identity analysis of the repABC of ATCC31749 C2 and C58 C2 also reveals that ATCC31749 C2_oriC shares 97.208% identity to that of C58.
A phylogenetic tree inferred from nucleotide sequences of the repABC operons of ATCC31749 C2, pAg31749a, pAg31749b, and C58 C2, pTiC58, pAtC58, pTiJ07, pAtJ07, pTiLBA4404, pAtLBA4404, pRtCB782a, pRtCB782b, pRtCB782c, pAtS4a, pAtS4b, pAtS4c, pAtS4e and pTiS4 finds ATCC31749 C2 and C58 C2 to be placed with symbiosis-related plasmids pRtCB782a and pRtCB782b. This tree suggests that ATCC31749 C2 and C58 C2 with the plasmids pRtCB782a and pRtCB782b diverged from the tumor-inducing plasmids pTiC58, pTiLBA4404, pTiJ07 and pTiS4 during the process of evolution. Hence, it is possible that ATCC31749 C2 may have a symbiotic/Ti plasmid ancestor. It can be deduced from this tree and the syntenic relationship data that ATCC31749 C2 may have evolved from an ancestral symbiosis/Ti plasmid which integrated into a duplicated copy of C1 (Figure 5a & 5b), rather than transfer of large number of genes from the C1 to the ancestral plasmid. The previous hypothesis by Slater et al., (6), require large number of genes transfer from the C1 to the ancestral symbiosis/Ti plasmid to form C2, and this may be difficult to achieve during the process of evolution. It is easier for the ancient symbiosis/Ti plasmid (or its fragments including its replication origin) to be integrated into a copy of the duplicated C1 to form C2, than large number of genes being transferred from C1 to the ancestral plasmid during the process of evolution. We therefore propose a possible pathway for evolution of the C2 of ATCC31749, whereby formation of C2 occurred by integration of fragments of an ancestral symbiosis/Ti plasmid into the duplicated copy of C1, followed by loss of genes (including some virulence genes) and inter-transfer of genes among the four replicons (Figure 5c). This is consistent with the syntenic relationship results which show that the C2 is more homologous to C1, and shares very few similar genes with the plasmids (Figure 5).???????
Plasmids
In figure 5b, plasmid pAg31749a is placed with pAt plasmids pAtC58, pAtLBA4404 and pAtJ07, whiles pAg31749b is placed with mutualistic symbiotic-related plasmid pRtCB782c of CB782. This tree also places ATCC31749 C2, pAg31749a and pAg31749b at different clades, confirming our result in figure S2 and therefore support our hypothesis that all the four replicons of ATCC31749 may have different origins of evolution. The replication origins of both pAg31749a and pAg31749b have the same pattern of the repABC replication system. Comparative analysis of the homologous protein sequences of repABC, reveals that pAg31749a and pAg31749b share 43% similarity for repA, 33.9% for repB and 58% for repC. BLASTN analysis of repABC and oriC of pAg31749a and pAg31749b, show that pAg31749a has 100% coverage and 99% identity with pAt in C58, whiles pAg31749b has 98% coverage and 82% identity with the mutualistic symbiosis-related plasmid (pRtCB782c) of Rhizobiumleguminosarum bv. trifolii CB782 (CB782). These results suggest that pAg31749a may have a common ancestral plasmid of origin of evolution with pAtC58, while pAg31749b may also have a common ancestral plasmid of origin of evolution with pRtCB782c in CB782. The gene function annotation data shows that the pAg31749a mainly contains secondary metabolites biosynthesis genes, and transport and metabolism related genes. Plasmid pAg31749b does not only contain secondary metabolites biosynthesis genes, but also primary metabolism genes including sugar, amino acids and lipids transport and metabolism related genes. The energy-producing and carbohydrate metabolism related genes of pAg31749b may also contribute to the synthesis curdlan.
Expression of curdlan biosynthesis genes
For analysis of the expressed genes of ATCC31749, 6 samples (3 replicates each for exponential (E) and stationary (S) growth phases) of ATCC31749 were used for RNA-seq analysis. Using FPKM value of 1 as a standard cut-off, any gene having FPKM value ≥ 1 was regarded as an expressed gene. Distribution of FPKM values across the 6 samples at E- and S-phases has been summarized in figure 6a. It was found that, 4567 genes were expressed. For analysis of differentially expressed genes (DEGs) between S- and E-phases, a fold change (fc) value ≥ 2 and q-value ≤ 0.05 were set as cut-off to select DEGs. It was found that, during the S-phase 207 genes were up regulated (File S4). Gene annotation data revealed that, genes that were down regulated in the S-phase are mainly involved in primary metabolism such as glycolysis, citric acid cycle, oxidative phosphorylation, protein, sugar, lipid and nucleic acid metabolism, cell replication, growth and development. On the other hand, genes that were up regulated in the S-phase are primarily involved in stress response activities, nitrogen transportation, signal transduction, and CPS and EPS biosynthesis, regulation and transport. The curdlan synthase operon (crdASC) and its regulatory-related genes were highly upregulated at the S-phase (Figure 6b and 6c, showing DEGs of fc ≥ 4). The synthase-dependent exopolysaccharide secretion genes TPR, β-Barrel, Tm-Barrel, stress response A/B-Barrel and their regulatory-related genes, and the signal molecule c-di-GMP-regulation associated genes which are necessary for curdlan biosynthesis and transport were upregulated at the S-phase. EPS/CPS biosynthesis and transport-related genes, nitrogen signalling cascade (NtrBC) and pyrophosphate (rrpP) were upregulated in the curdlan biosynthesis stage (S-phase). Some regulatory genes such as cAMP phosphodiesterase (cpdA) and oxygenase (FixL,FixJ) that are vital for improvement of curdlan biosynthesis were also upregulated.
At the E-phase, genes that were upregulated are mostly primary metabolism related genes for growth and development (such as glucose-6-phosphate isomerase, sucrose-phosphate hydrolase, UTP-glucose-1-phosphate uridylyltransferase, UDP pyrophosphate, succinate dehydrogenase, argininosuccinate synthase, adenylosuccinate synthetase, GTP binding proteins, acetyl-CoA acetyltransferase and synthase, fructose-1,6-bisphophate aldolase, pyrophosphate-fructose-6-phosphate 1-phosphotransferase, malate dehydrogenase, glutamine synthetase, glutamine amidotransferase, phosphoglycerate kinase, pyruvate dehydrogenase), and some regulatory and energy providing-related genes (such as AMP and GMP synthase diguanylate cyclases, diguanylate phosphodiesterases; nitrogen fixation regulatory genes; EPS production negative regulators). Based on our ATCC31749 genome and transcriptome data, the process for curdlan biosynthesis and regulation with extracellular raw materials such as glucose or sucrose are summed up in Figure 6d.
Curdlan acts in defence of ATCC 31749 cells
To study the functions of curdlan to the ATCC31749 cells, three different strains of ATCC31749 cells that have been grown to E-phase and S-phase. The strains include ATCC31749 (Curdlan-producing ATCC31749, wild type), ATCC31749-?crdR (ATCC31749 mutant of crdR knockout) with dramatically reduced curdlan synthesis [38] and ATCC31749-?crdR+pBQcrdR (ATCC31749?crdR mutant that has been complimented by crdRinpBQcrdR) were used to analyze the protective ability of curdlan on ATCC31749 cells against stress conditions. The stress conditions investigated were high temperature (42?C), UV light and acidic conditions. We found that, the number of colonies that grew on the agar plates for all three strains at the curdlan producing stage (S-phase) were significantly higher than their counterparts at the E-phase for all conditions studied. This is possible because at the cell growth stage (E-phase), lower or no curdlan has been produced. When the number of colonies of the three strains at any growth stage were compared, it was found that, the wild type strain (ATCC31749) had significant higher number of colonies that grew than the other two strains (Figure 7). The ?crdR mutant strain (ATCC31749-?crdR), which lacks crdR gene that regulates curdlan biosynthesis, produced the lowest number of colonies for all conditions studied. This suggest that curdlan acts in defense of ATCC31749cells by forming a protective sheath that cover the cells surfaces. It can therefore be inferred that, curdlan serves a protective barrier between the cells surfaces and their immediate environment.
Discussion
ATCC31749 is of particular biological interest because of its ability to synthesize curdlan which is of high importance to food and pharmaceutical industries. Therefore, an insight into the complete genome of ATCC31749 will be helpful for the scientific community and industrial producers of curdlan. Genomes of organisms of the order Rhizobiales are highly enriched with genes for regulation of pathogenic or symbiotic processes, secondary metabolites, transport and stress-related systems such as EPS for self-protection (13). These genomes contain single or multiple chromosomes (some of which are linear) and plasmids (6). The genome of ATCC31749 contains two chromosomes, two plasmids and 14 predicted genomic islands. The two plasmids of ATCC31749 are highly enriched with symbiotic-related genes such as nodulation genes (nodN, nodT, nolG, nolR) that encode proteins involved in the biosynthesis and transport of nodulation factors which induce nodule organogenesis in most rhizobia. Other symbiotic-related genes present include nitrogen-fixation genes (fixL, fixG, fixH, nifR, nifH, NtrABC). The presence of symbiotic-related genes in ATCC31749 plasmids, pAg31749a and pAg31749b, supports available literatures that states that in Agrobacteria, mutualism and symbiosis-related genes are basically encoded on the transmissible plasmids or mobile gene islands (Symbiotic Islands) which permits the conversion of non-symbiotic organisms into nitrogen-fixing plant endosymbionts and vice versa. The organization of these symbiotic genes clustered within mobile islands or plasmids are not only necessary for genome expansion but also specifies the genomic expansion by both gene duplication and accessory nature of genes acquired by horizontal gene transfer [39-41]. The primary chromosome of ATCC31749 which is the circular chromosome contains a putative origin of replication (oriC) that has a 92.252% identity to the known oriC of C58. The secondary (linear) chromosome of ATCC31749 has a plasmid-type replication system of the same type found on C58 linear chromosome and shares 97.208% identity to that of C58. Furthermore, the ATCC31749 genome nucleotide sequence shares 98% identity (at a coverage of 85%) with the genome nucleotide sequence of C58, indicating that the ATCC31749 genome resembles more closely to genomes of Agrobacteriumfabrum (also called A. tumefaciens) group than the genomes of non-pathogenic Agrobacterium species, although ATCC31749 is not virulent. It is therefore possible that the ancestors of ATCC31749 might have lost their virulence genes during the evolution process.
In multichromosomal bacteria, multichromosomes may be necessary for effective and efficient completion of genome replication and segregation in less time. The fewer the replication forks, the lesser the breakages and hence, the lesser the formation of dimers resulting from repair of broken replication forks. This improves genome maintenance and chromosome stability during rapid growth [42]. Furthermore, formation of a secondary chromosome in ATCC31749 may serve as an alternative reservoir for newly acquired genes and as a backup gene copy for some essential genes (as seen in figure 5a). Multichromosomes that are found in diverse forms of bacteria, evolved through different processes. These processes include chromosome excision into different replicons, duplication of genome following independent structure diversification, mutation of multicopy chromosomes, unequal chromosomal division, horizontal gene transfer and transformation of a plasmid into a chromosome [6, 43-46]. In Rhizobiales, two types of evolutionary paths have been proposed for chromosome evolution. One of these evolutionary mechanisms is the integration of the ancestral plasmid into the primary chromosome as seen in Bradyrhizobium strains [47]. The other mechanism for chromosome evolution is the transfer of chromosomal genes from primary chromosomes to ancestral plasmids resulting into formation of secondary chromosomes (a plasmid-based mechanism for formation of secondary chromosomes) as seen in Proteobacteria. The plasmid-based mechanism for formation of secondary chromosomes require large number of genes to be transferred from the primary chromosome to the ancestral plasmid. This may not be easy to be achieved than just the whole ancient symbiosis/Ti plasmid or partial fragments of the ancient symbiosis/Ti plasmid including its replication origin integrating into a duplicated copy of the ancient chromosome to form an ancestral secondary chromosome. The repABC genes involved in replication, copy number control, partitioning and stability maintenance of replication of repABC-based replicon are highly phylogenetically distinct. Therefore, a phylogenetic tree inferred from repABC is a good source of data for evolution studies. Our repABC phylogenetic tree finds ATCC31749 C2 and C58 C2 to be placed with mutualistic symbiosis-related plasmids pRtCB782a and pRtCB782b, which might have diverged from tumor-inducing plasmids pTiC58, pTiLBA4404, pTiJ07 and pTiS4 (Figure 5b). It is therefore possible that, ATCC31749 C2 may have evolved from an ancestral symbiosis/Ti plasmid. Furthermore, the syntenic relationship tBLASTx graphical comparison of the chromosomes (C1, C2) with the plasmids (pAg31749a and pAg31749b) also suggest that, the secondary chromosome shows very high similarity to the primary chromosome (Figure 5a) with BLASTN identity of 86.6%, inferring to a possibility that C2 may have evolved from duplication of C1. This is not consistent with the hypothesis of previous literature (6) which states that secondary chromosomes of Agrobacterium biovar I/III originated from ancestral plasmids to which chromosomal genes have been transferred to since the secondary chromosomes contain plasmid-type replication origin repABC. Although, this hypothesis is a possibility, it is also possible that secondary chromosomes may originate from a duplicated copy of the primary chromosome to which the repABC fragments with other fragments of the ancestral plasmid have been integrated into. We therefore propose that accumulation of ancient plasmid and/or plasmid genes into the duplicated C1 led to formation of the secondary chromosome (C2) in ATCC31749.Our proposed pathway for formation of C2 by integration of fragments of an ancestral symbiosis/Ti plasmid, followed by loss/gain of genes including virulence genes, and transfer of genes among the replicons and linearization of the ancestral C2 during the long history of evolution has been summarized in figure 5c. The features of the ends of the linear chromosome are consistent with the presence of telomere-like structures which play crucial roles in the integrity and stability of linear chromosome, preventing end-to-end fusions and DNA loss during replication [48-50]. These features confirm our result that the secondary chromosome is a linear chromosome. The same properties are found on the C58 secondary chromosome which is a linear chromosome.
EPS production is crucial for bacteria survival during unfavourable environmental conditions. Exopolysaccharide biosynthesis processes include synthesis of sugar nucleotide precursors (a rate-limiting step for exopolysaccharide), assembly of the repeating unit, and polymerization and transportation to the extracellular environment where the EPS performs its function of protection or attachment. As a protective mechanism, C58 produces extracellular β-1,2-glucan (a CPS) which is transported via the ABC transporter dependent pathway to the extracellular where it is linked to the C58 cell surface (33) enabling the cells to easily attach to host plants. The ABC transporter dependent pathway is ATP-dependent and requires high genetic composition (more genes) such as GTs, OPX, PCP and poly-kdo-linker. Furthermore, the degree of polymerization of β-1,2-glucan is determined by the C-terminal domain of Cgs gene (NdvB and ChvB) [51] which facilitates the four enzymatic reactions: initiation, elongation, phosphorolysis and cyclization [52] of β-1,2-glucan production, which is accomplished by multiple synthase genes. Unlike C58 and other Agrobacterium symbionts, ATCC31749is independent of any plant host and therefore has evolved to maintain a manageable genome size with less gene expression requirements to survive adverse conditions such as nitrogen-limited conditions. Over the period of evolution, ATCC31749has adapted to a transport system (Synthase-dependent transport pathway) with less genetic requirements. Unlike the polymerization of β-1,2-glucan of C58, the polymerization of curdlan (a β-1,3-glucan) as well as the transportation process of the synthase-dependent transport pathway is performed by a single synthase gene. Synthase dependent pathway is usually used for the production and assembly of homopolymers such as curdlan and cellulose that require only one type of sugar precursor (2). Apparently, ATCC31749 may have evolved and adapted to the production of curdlan not only as a protective measure to withstand stress and adverse conditions (Figure 7), but also to by-pass the high genetic requirement of symbiosis. Furthermore, because curdlan is a water-insoluble and alkali-soluble biopolymer, ATCC31749 may have adapted to the synthesis of curdlan over the process of evolution and selection, as an energy reservoir and medium of attachment to solid surfaces for better survival when in water medium.
Conclusion
The genome of ATCC31749is composed of two chromosomes and two plasmids. Genes that are responsible for housekeeping activities and basic life processes are located on the primary chromosome, although curdlan biosynthesis genes are located on the secondary chromosome. The secondary chromosome of ATCC31749 is derived from a duplicated copy of the primary chromosome to which fragments (including the origin of replication) of an ancestral symbiosis/Ti plasmid has integrated into. ATCC31749 is found to be grouped with pathogenic Agrobacterium species and its genome resembles more closely to the genomes of pathogenic Agrobacterium species than non-pathogenic Agrobacterium species. A. fabrum str. C58 is the closest related Agrobacterium species to ATCC31749 and they may have a common pathogenic/symbiotic Rhizobiales genetic ancestor. ATCC31749 is an independent Agrobacterium species that produces curdlan as a protective glucan to protect itself against unfavourable conditions.
Supplementary data
The following files were uploaded as "Supplementary Files" during the manuscript submission process.
Figure S1: Phylogenetic tree inferred from 57 Agrobacterium/Rhizobium group species by Maximum likelihood analysis.
Figure S2: Phylogenetic trees inferred from OriC DNA nucleotide sequences of the primary and secondary chromosomes of 41 multichromosomal bacteria species.
File S1: Genome sequence of ATCC31749.
File S2: List of 247 highly orthologous genes of the 57 species.
File S3: Genome annotation ofATCC31749.
File S4: Differentially expressed genes of ATCC31749.
Authors contributions
“Conceptualization, Chun Lin and Zichao Mao; Data curation, Huifang Sun and Zhengjie Liu; Formal analysis, Rex Frimpong Anane , Huifang Sun and Lamei Zhao; Funding acquisition, Zichao Mao; Investigation, Rex Frimpong Anane, Huifang Sun and Lamei Zhao; Methodology, Chun Lin and Lamei Zhao; Project administration, Zichao Mao; Resources, Chun Lin and Zichao Mao; Supervision, Zhengjie Liu; Validation, Rex Frimpong Anane, Chun Lin and Zichao Mao; Visualization, Rex Frimpong Anane, Zhengjie Liu and Zichao Mao; Writing – original draft, Rex Frimpong Anane; Writing – review & editing, Rex Frimpong Anane and Zichao Mao.”
Conflicts of interest
The authors declare that there are no conflicts of interest.
Funding information
This research was funded by CHINESE NATURE SCIENCE FOUNDATION, grant numbers 31360085; 31170057. The funding was obtained by the author Zichao Mao. The funders did not employ anyone to participate in the research.
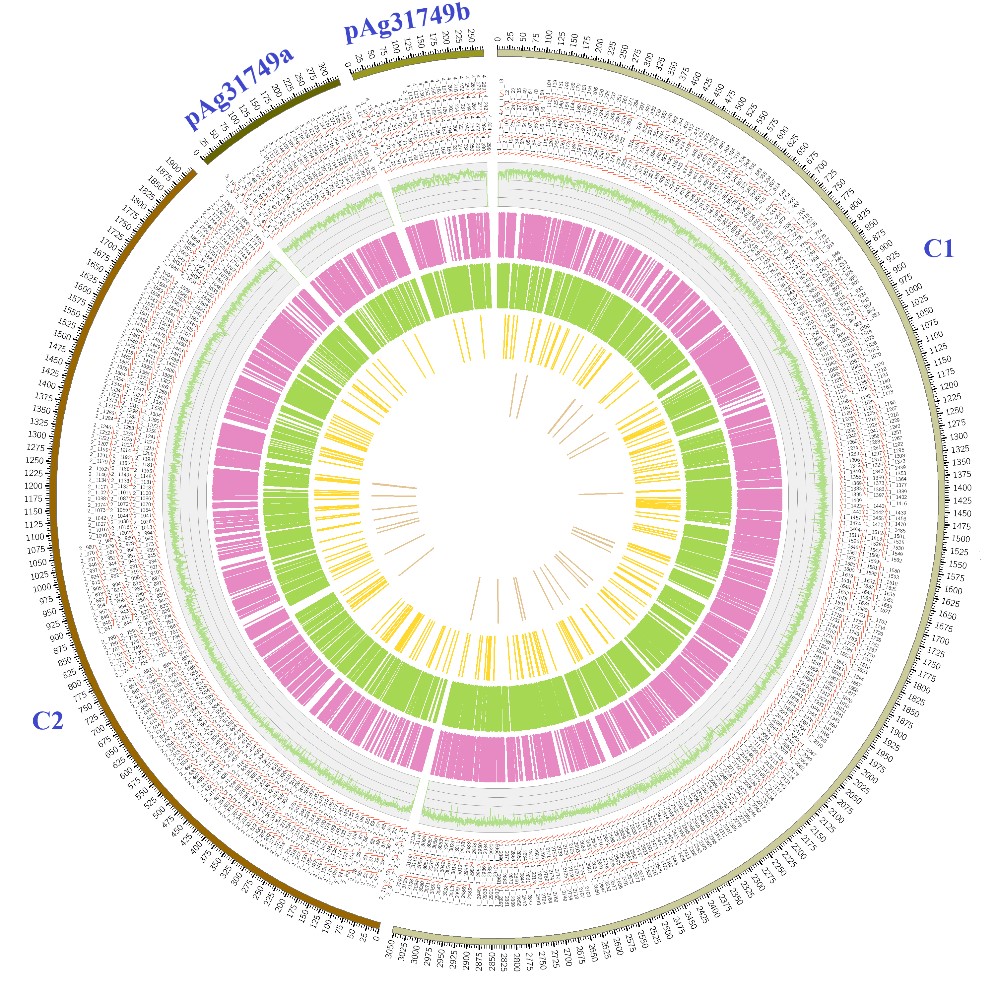
Figure 1: Genome features of Agrobacterium sp. ATCC 31749. The genome consists of two small circular plasmids (pAg31749a and pAg31749b), one large circular chromosome (C1) and one linear chromosome (C2). Their predicted genes are indicated outside the circles 1 and 2 (from the outer to the inner) with kb as genomic length unit. The predicted tRNAs and transposons are shown in circles 4 and 5, while the 3rd circle depicts the GC content.The annotated proteins encoded by the leading and the lagging strand (inner circles 6 and 7) are marked with different colors
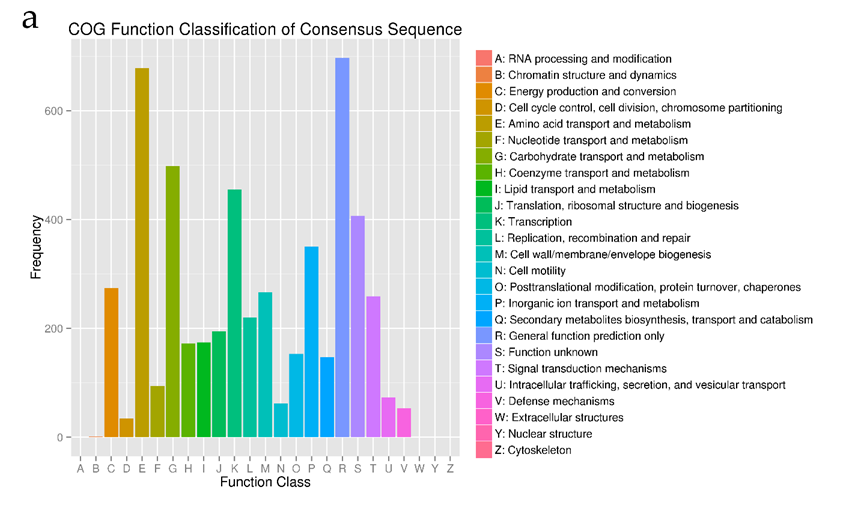
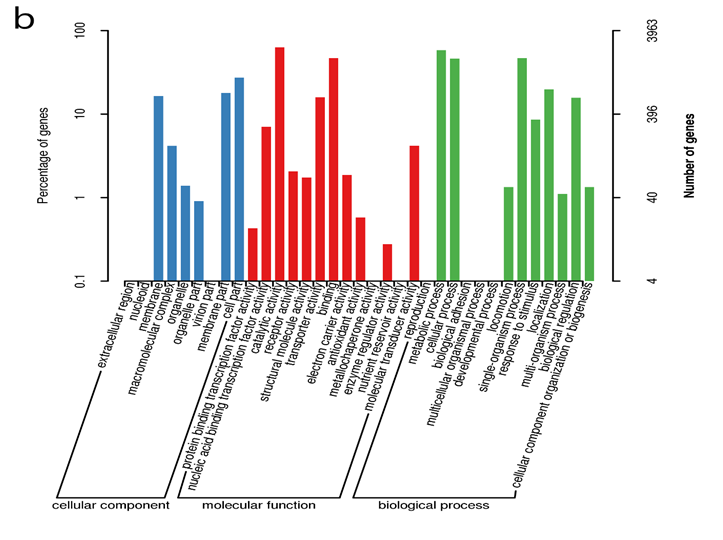
Figure 2: Classification for gene function for Agrobacterium sp. ATCC 31749. (a) Conserved Orthologous Genes (COG) Classification showing that high frequency of genes function in transport and metabolism (E-I, P-Q). (b) Gene Ontology (GO) classification showing that majority of the genes are involved in molecular function and biological processes
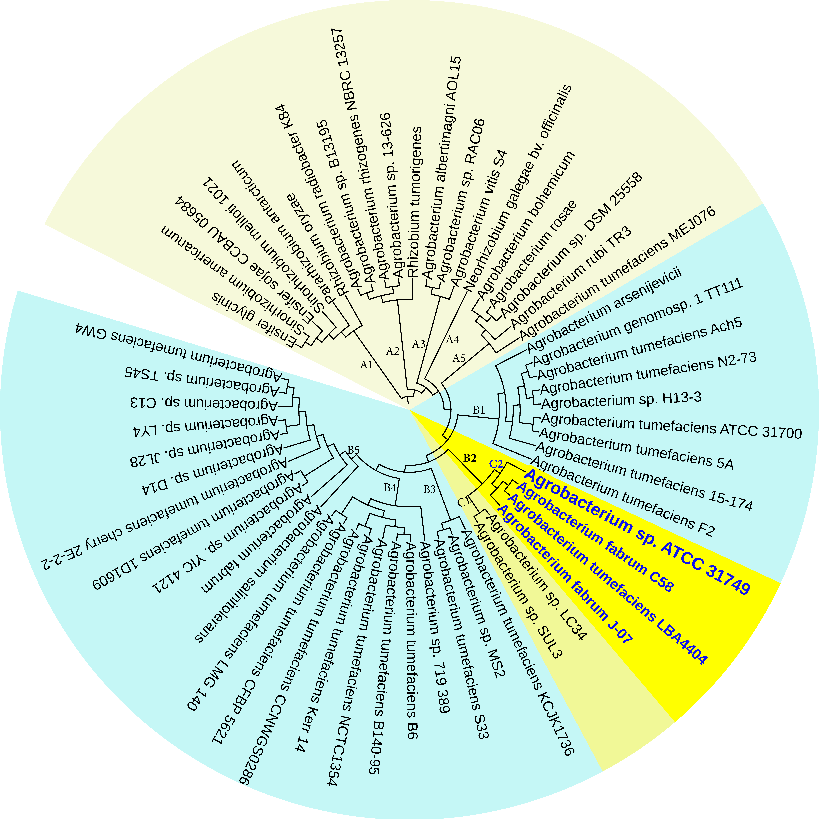
Figure 3: The NJ phylogenetic tree highlighting the position of Agrobacterium sp. ATCC 31749 relative to other 56 species within the Agrobacterium/Rhizobium group. The tree was inferred from 247 protein sequences of highly orthologous genes. This phylogenetic tree suggests that C58 is the closely related species to ATCC31749 and that the genome of ATCC31749 is very similar to those of Agrobacterium fabrum group although ATCC31749 is not pathogenic.
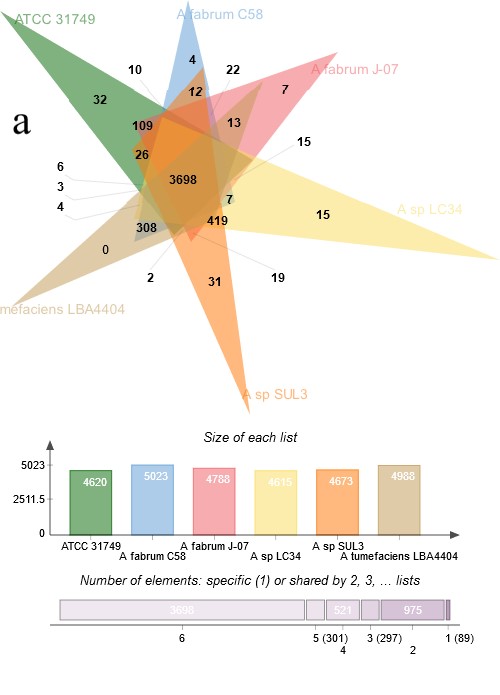
Figure 4:Comparative genomics between ATCC31749 and other species. (a) Venn diagram depicting orthologous cluster of genes shared between ATCC31749and the closest related species (C58, J-07, LBA4404 LC34 and SUL3) in the clade “B2” of figure 3. The numbers depict the number of orthologous clusters of genes shared between the six species. The bar graph below the venn diagram shows the size of gene clusters in each species, whiles the diagram below the bar graph shows the number of specific or shared genes by 1, 2, 3, 4, 5 or 6 species.
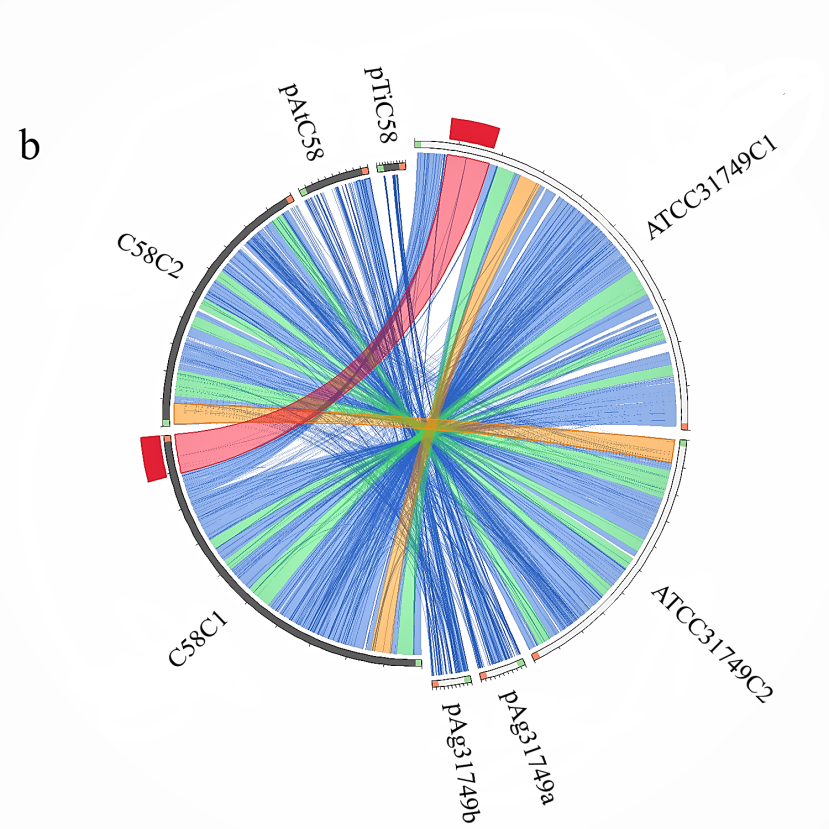
Figure 4:Comparative genomics between ATCC31749 and other species. (b)Circos plot showing syntenic genes relationship between the replicons of ATCC31749 and C58. ATCC31749C1 is highly homologous to C58C1, whiles ATCC31749C2 is more homologous to C58C2 with some genes being mapped to pTiC58. pAg31749a is highly mapped to the plasmid pAtC58, whiles pAg31749b is highly mapped to C58C2 with very few fragments being mapped to pAtC58 and pTiC58.
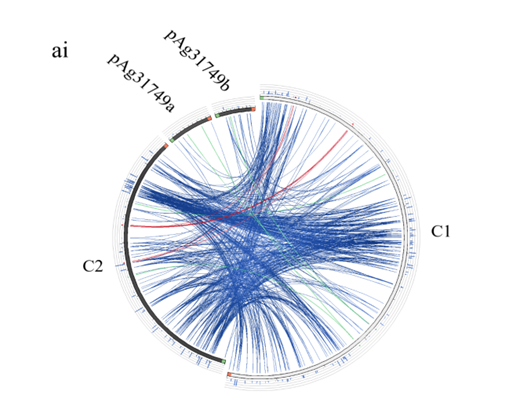
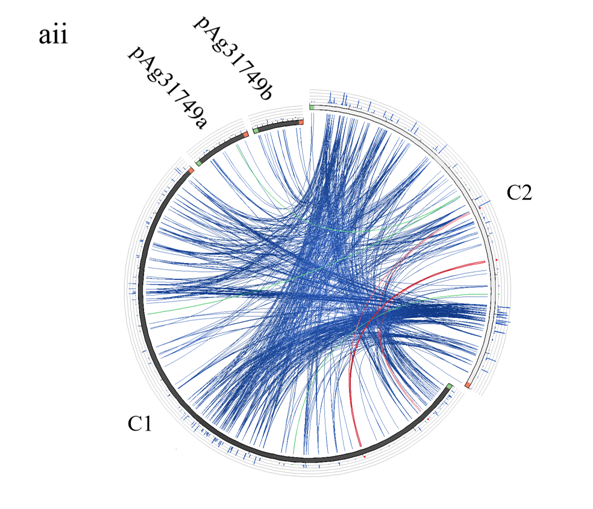
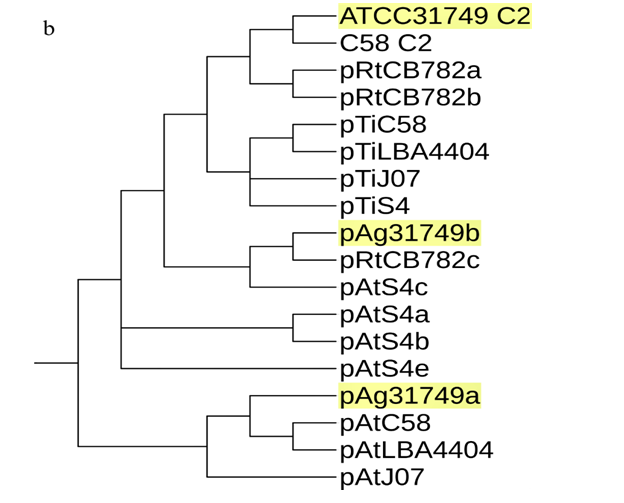
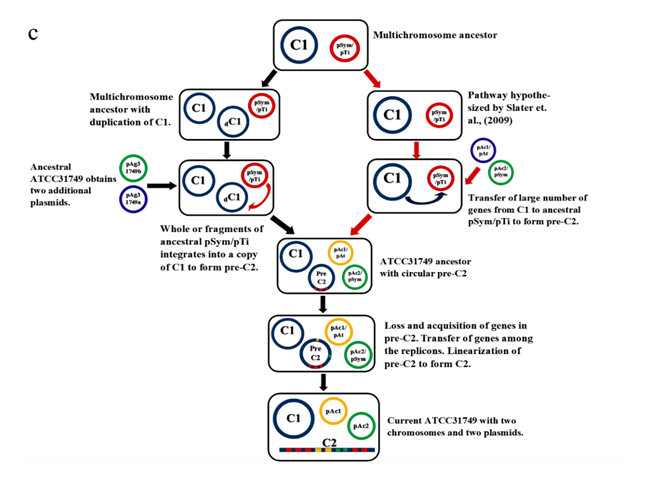
Figure 5: Evolution of the secondary chromosome of ATCC31749. (ai) Circos plot showing similarities (tBLASTx) between the primary chromosome (C1) and the other replicons of ATCC31749. (aii)Circos plot showing similarities (tBLASTx) between the secondary chromosome (C2) and the other replicons of ATCC31749. The threshold for connecting lines was set at e-value ≤ 10−10, with line colors reflecting the ratio of actual tBLASTxbitscore to the maximal score (using ‘score/max’ ratio coloringof blue ≤ 0.25, green ≤ 0.50, orange ≤ 0.75, red > 0.75). The outer histogram counts how many times each color has hit the specific part of the sequence and uses an equivalent coloring scheme. (b) Phylogenetic tree inferred from repABC genes highlighting the origin of C2 of ATCC31749. (c) The proposed possible pathway for the evolution of the secondary chromosome (C2) of ATCC31749 (shown in black coloured arrows).
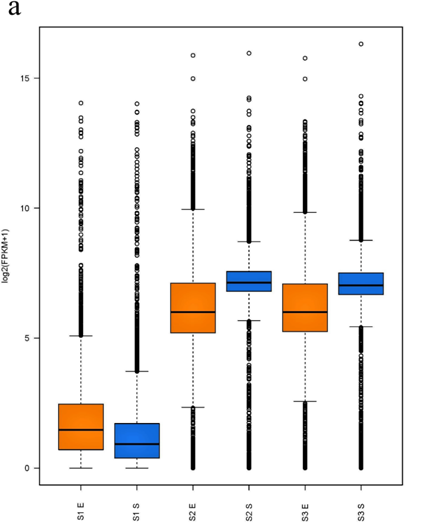
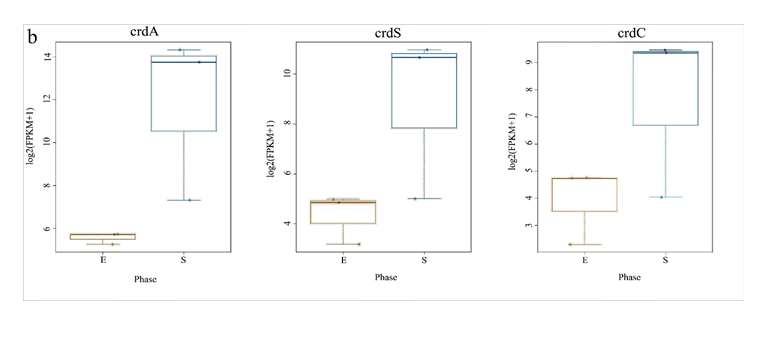
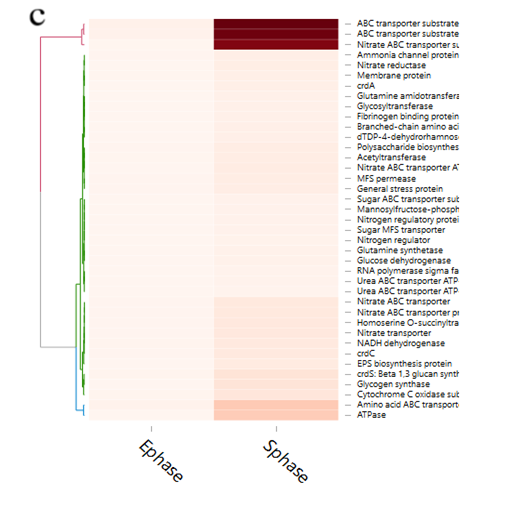
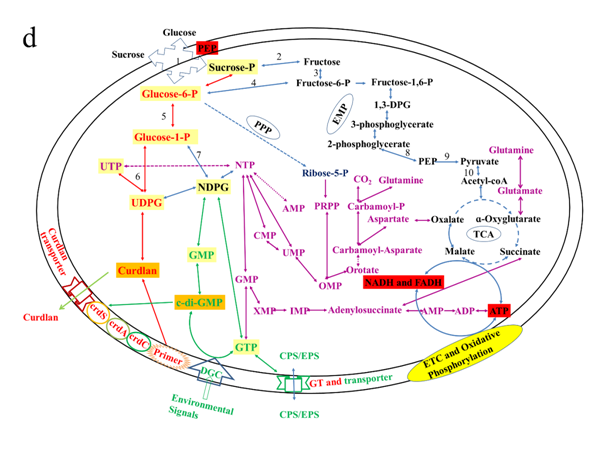
Figure 6: Expression of curdlan biosynthesis genes. (a) Distribution of FPKM values across the 6 samples (S1E – S3S) at E- and S-phases. Samples from the same phase are shown in the same color. E-phase in orange and S-phase in blue. (b) FPKM distributions at E-phase and S-phase for crdA, crdSand crdC genes (curdlan synthesis operon). These are genes that are known to be highly expressed at S-phase, displayed as box-and-whiskers plots. (c) Average expression level (RPKM) for curdlan biosynthesis related genes across all samples at E- and S-phases. Curdlan biosynthesis operon and its regulatory-related genes, and stress-response related genes were highly upregulated at the S-phase. Red rectangles present genes showing specific high expression levels. The deeper the red color the higher the expression level. (d) Biosynthetic pathway and regulatory networks for curdlan in ATCC31749. Curdlan biosynthesis pathway is shown in red words with yellow background. NTP biosynthesis pathway for providing energy in curdlan biosynthesis is in purple words. C-di-GMP biosynthesis pathway for regulating curdlan biosynthesis is in green words with yellow background. Primary metabolism such as glycolysis and TCA are in black words. 1. PTS system, 2. phosphosucrose hydrolase, 3. Fructose kinase or hexoses kinase, 4. phosphoglucoisomerase, 5. Phosphoglucomutase, 6. UTP-glucose-1-phosphate uridylyltransferase, 7. sugar nucleotide synthase, 8. Enolase, 9. pyruvate kinase, 10. 2-oxoacid ferredoxin oxidoreductase.
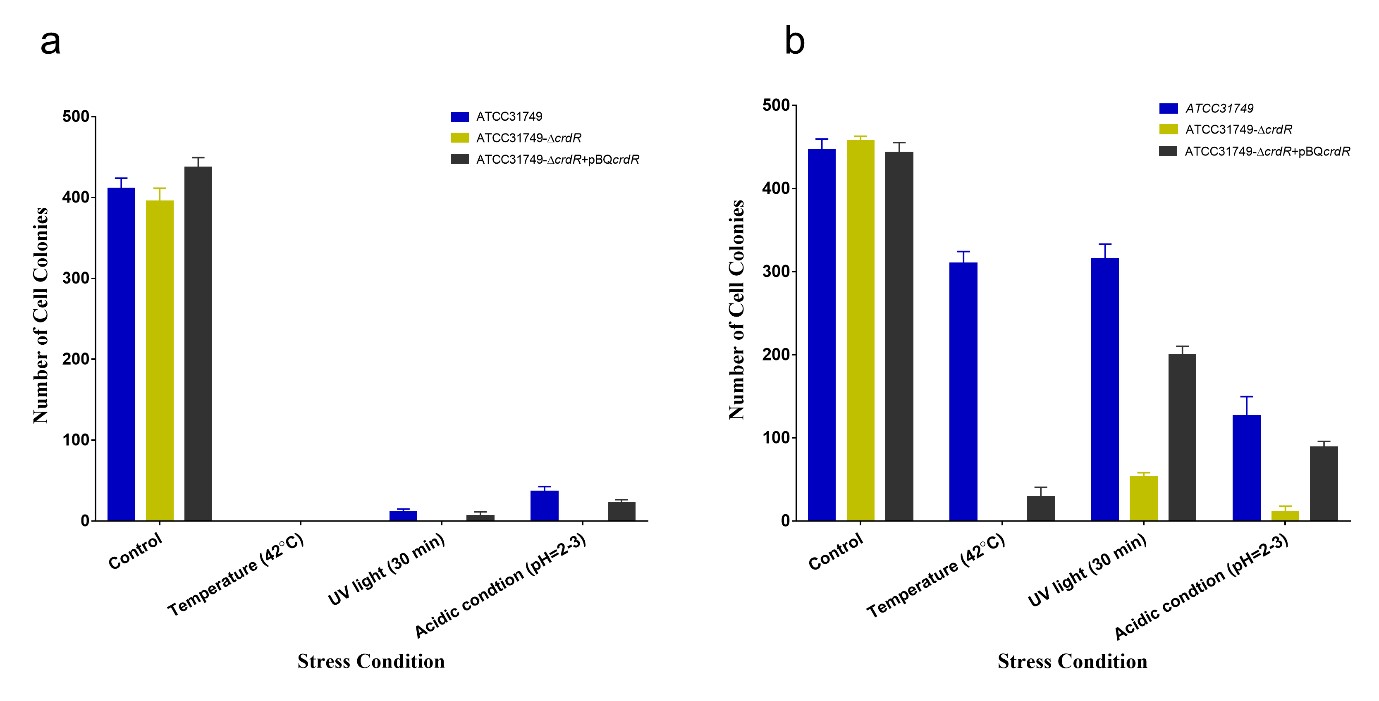
Figure 7: Protective ability of curdlan on ATCC31749cells. “b” represents the S-phase and “a” represents the E-phase. The number of cell colonies that grewon the agar plates for each cell type at the S-phase were significantly higher than the colonies that grew at the E-phase. At each growth phase, the wild type strain (ATCC31749) produced the highest number of colonies followed by the ATCC31749-?crdR+pBQcrdR, the mutant strain that has been transformed with crdRgene. The ATCC31749-?crdR mutant strain produced the lowest number of cell colonies at both growth phases, indicating that curdlan serve as a protective cover for the ATCC31749 cells.
Supplementary data

Mao Figure S1 - tree Final

Mao Figure S2a

Mao Figure S2b
|
Characteristic |
Agrobacterium sp. ATCC 31749 |
|
Genome size (bp) |
5,566,198 |
|
G+C content (%) |
58.9 |
|
Number of chromosomes |
2 |
|
Estimated coverage (%) |
100 |
|
Number of plasmids |
2 |
|
Number of protein-coding genes |
5591 (Primary chromosome, 3142; Secondary chromosome, 1811; pAg31749a, 331; pAg31749b, 307) |
|
Size of protein-coding genes (bp) |
4861713 |
|
Mean length (bp) |
869 |
|
Number of genomic islands |
14 |
|
Total size of islands (bp) |
228495 |
|
Number of hypothetical proteins |
1,150 |
|
Number of pseudogenes |
21 |
|
Number of prophages |
3 |
|
Length of prophages (bp) |
(1) 58795, (2) 26930, (3) 58962 |
|
Repeats |
245 |
|
rRNA operon |
4 |
|
tRNA |
33 |
|
mRNA |
5591 |
|
Coding density (%) |
87.3 |
|
Number of CRISPR elements |
1 |
Table 1:Genome assembly statistics
Citation: Anane RF, Lin C, Sun H, Zhao L, Liu Z, et al. (2019) Complete Genome Sequence of Agrobacterium sp. ATCC 31749 and Insights into its Curdlan Biosynthesis. CurrTrends GenetMicrobiol: CTGM-100001
