
Current Trends in Genetics and Microbiology
Review Article
The Role of Genetic Mutations in Genes CACNA1C, CACNB2, SCN1B, KCNE3, KCND3, SCN10A, HEY2, SCN5A, GPD1L in Brugada Syndrome
Asadi S* and Yousefi R
Division of Medical Genetics and Molecular Pathology Research, Harvard University, Boston Children's Hospital, USA
*Corresponding author: Shahin Asadi, Division of Medical Genetics and Molecular Pathology Research, Harvard University, Boston Children's Hospital, USA.
Citation: Asadi S and Yousefi R (2020)The Role of Genetic Mutations in Genes CACNA1C, CACNB2, SCN1B, KCNE3, KCND3, SCN10A, HEY2, SCN5A, GPD1L in Brugada Syndrome. Curr Trends Genet Microbiol: CTGM-100002
Received date: 29 July, 2020; Accepted date: 03 August, 2020; Published date: 07 August, 2020
Abstract
Brugada syndrome is a genetic disorder that causes the heart rhythm to become abnormal. In fact, this syndrome can lead to an irregular heartbeat in the left ventricle, also known as ventricular arrhythmia. Signs and symptoms of ventricular arrhythmias, including sudden death due to cardiac arrest, can occur from birth to late puberty. Sudden death in people with Brugada syndrome usually occurs around the age of 40. The genetic form of Brugada syndrome is most often caused by a defect in the SCN5A gene but other genes can be involved, too. It can be inherited from just one parent. However, some people develop a new defect of the gene and don't inherit it from a parent. This genetic defect causes heart muscles cells to handle sodium abnormally, which can then lead to abnormal heart rhythms.
Sometimes you may have Brugada syndrome but it's dormant and doesn't cause any problems. However, some medicines such as antidepressants and antipsychotics, illicit drugs, conditions that cause fever, and electrolyte problems can unmask the syndrome. Sometimes people may appear to have Brugada syndrome based on an electrocardiogram but don't have the disease itself. This is called a Brugada ECG pattern and may not pose a risk if the condition is temporary and doesn't cause symptoms or dangerous heart rhythms.
Keywords: Brugada Syndrome; Heart Disorder; Genetic Mutations; SCN5A; CACNA1C; CACNB2; SCN1B; KCNE3; KCND3; SCN10A; HEY2; GPD1L Genes
Overview of Brugada Syndrome
Brugada syndrome is a genetic disorder that causes the heart rhythm to become abnormal. In fact, this syndrome can lead to an irregular heartbeat in the left ventricle, also known as ventricular arrhythmia. If not treated or controlled properly, an irregular heartbeat can cause fatigue, seizures, difficulty breathing or cardiac arrest. These complications usually occur when a person with Brugada syndrome rests with sleep [1](Figure 1).
Clinical signs and symptoms of Brugada syndrome
The clinical signs and symptoms of Brugada syndrome usually appear in adulthood, although they can appear at any time during life. Signs and symptoms of ventricular arrhythmias, including sudden death due to cardiac arrest, can occur from birth to late puberty. Sudden death in people with Brugada syndrome usually occurs around the age of 40. This condition can include some cases of Sudden Infant Death Syndrome (SIDS), which is one of the leading causes of death in infants less than one-year-old. In addition, Unknown Sudden Death Syndrome (SUNDS) is a condition caused by an unexpected heart disorder in young adults, usually at night, and researchers believe that SUNDS Syndrome and Brugada Syndrome are similar disorders [2](Figure 2).
Etiology of Brugada syndrome
Researchers have divided Brugada Syndrome into 6 different types: Brugada Syndrome Type 1 (BS1), Brugada Syndrome Type 2 (BS2), Brugada Syndrome Type 3 (BS3), Brugada Syndrome Type 4 (BS4), Brugada Syndrome Type 5 (BS5) and Brugada Syndrome Type 6 (BS6) [1-6].
Each type of Brogada syndrome is caused by mutations in different genes. For example, Brugada type 1 syndrome is caused by a mutation in the SCN5A gene, which is located in the short arm of chromosome 3 as 3p22.2. Brugada type 2 syndrome is caused by a mutation in the GPD1L gene, which is located in the short arm of chromosome 3 as 3p22.3 [1,3](Figure 3).
Brugada syndrome type 3 is caused by a mutation in the CACNA1C gene, which is located in the short arm of chromosome 12 as 12p13.33. Brugada syndrome type 4 is caused by a mutation in the CACNB2 gene, which is located in the short arm of chromosome 10 as 10p12.31-p12.33. Brugada type 5 syndrome is caused by a mutation in the SCN1B gene, which is located on the long arm of chromosome 19 as 19q13.11. Brugada type 6 syndrome is caused by a mutation in the KCNE3 gene, which is located in the long arm of chromosome 11 as 11q13.4, and the KCND3 gene, which is located in the short arm of chromosome 1 as 1p13.2. In addition, mutations in the SCN10A gene, which is located in the short arm of chromosome 3 as 3p22.2, and mutations in the HEY2 gene, which is located in the long arm of chromosome 6, as 6q22.31, are also implicated in Brugada syndrome. However, the most common mutated gene in Brugada syndrome is the SCN5A gene, which accounts for about 30% of cases of Brugada syndrome, leading to Brogada syndrome type 1. The SCN5A gene provides the instructions for creating a sodium channel, which usually transports sodium atoms to the heart muscle cells. This type of ion channel plays an important role in maintaining the normal rhythm of the heart. Thus, mutations in the SCN5A gene alter the structure or function of the heart's sodium ion channel, reducing the flow of sodium ions to cells. Thus, impaired transport of sodium ions to the heart leads to abnormal heart rhythms in Brugada syndrome [1,4](Figure 4,5).
It is worth noting that mutations in other genes mentioned can cause less than 2% of cases of Brugada syndrome. For example, mutations in the CACNA1C and CACNB2 genes disrupt the calcium channel in the heart, which in turn disrupts the normal rhythm of the heart. Mutations in the KCNE3 and KCND3 genes also interfere with the release of potassium ions from the sodium-potassium pump, and mutations in the GPD1L gene disrupt the glycerol-3-phosphate dehydrogenase peptidase enzyme, all of which lead to abnormal rhythm. Are involved. The SCN1B gene, like the SCN5A gene, plays a role in the sodium channel of heart muscle cells [1,5](Figure 6-12).
Brugada syndrome follows an autosomal dominant inheritance pattern. Therefore, to produce this syndrome, only one copy of the mutant genes SCN5A, SCN1B, GPDL1, KCND3, KCNE3, CACNB2, CACNA1C (either parent) is required, and the chances of having a child with Brugada syndrome in this case, it is 50% for each possible pregnancy. It is worth noting that Brugada syndrome may also occur sporadically due to new gene mutations, in which case it will not follow any inherited pattern [1,6,7](Figure 13,14).
Frequency of Brugada syndrome
The exact prevalence of Brugada syndrome is unknown, although it is estimated to be around 5 in 10,000 people worldwide. Brugada syndrome is more common among Asian ethnic groups, especially people of Japanese and Southeast Asian descent. Brugada Syndrome affects both men and women. However, Brugada syndrome is 8 to 10 times more common in men than women. Researchers believe that the sex hormone testosterone, which is secreted in high levels in men, is the reason for the higher frequency of Brugada syndrome in men than women [1,8,9](Figure 15).
Diagnosis of Brugada Syndrome
Brugada syndrome is diagnosed based on the clinical signs and symptoms of the patient using specialized cardiovascular tests. An electrocardiogram can also be helpful in diagnosing Brugada syndrome. However, the most definitive way to diagnose Brugada syndrome is to test molecular genetics for these genes to check for possible mutations [1,10](Figure 16).
Brugada Syndrome Treatment Pathways
The treatment and management strategy of Brugada syndrome is based on the clinical findings of the patients. The most appropriate way to control this disease is early diagnosis and intervention to seek timely treatment in order to alleviate the suffering of patients. Regular use of cardiovascular drugs with a doctor's prescription will also be very helpful. In some cases, heart surgery can be effective in improving the condition of people with Brugada syndrome. Genetic counseling is also essential for all parents who want a healthy child ]1,11].
History of Brugada Syndrome
Brugada Syndrome was first discovered in November 1992 by two physicians, Brugada P and Brugada J [1,12](Figure 17).
Conclusion
Brugada syndrome is a genetic disorder that can causes a dangerous irregular heartbeat. In many cases, a defect in the SCN5A and other genes causes the genetic form of this condition. When this defect occurs, it may cause a ventricular arrhythmia. This is a type of irregular heartbeat. When this happens, the lower chambers of your heart (ventricles) beat irregularly and prevent blood from circulating properly in your body. This can be dangerous and may result in fainting or even death, especially during sleep or rest. The disease has been known as sudden, unexplained nocturnal death syndrome because people with it can often die in their sleep. Currently, there is no cure for Brugada syndrome. But, there are ways to protect people from the dangerous consequences of the disease. An implanted cardioverter defibrillator (ICD) can help prevent sudden death related to Brugada syndrome. When this device detects the start of an arrhythmia, it will either try to stop it with pacing or deliver a shock to reset it back into its regular rhythm. Medicines may also help prevent arrhythmias. Another potential treatment option may be a cardiac ablation. In this procedure, energy is directed to destroy the area of heart tissue causing the dangerous arrhythmia. Discuss all options with a qualified healthcare provider [1,12].
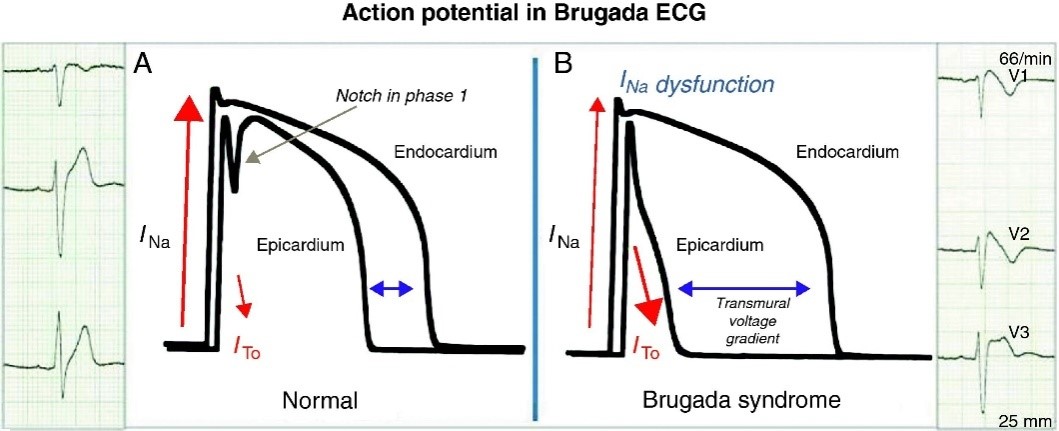
Figure 1: Line chart of electrocardiogram of the heart with sodium ion flow in the cell [1].
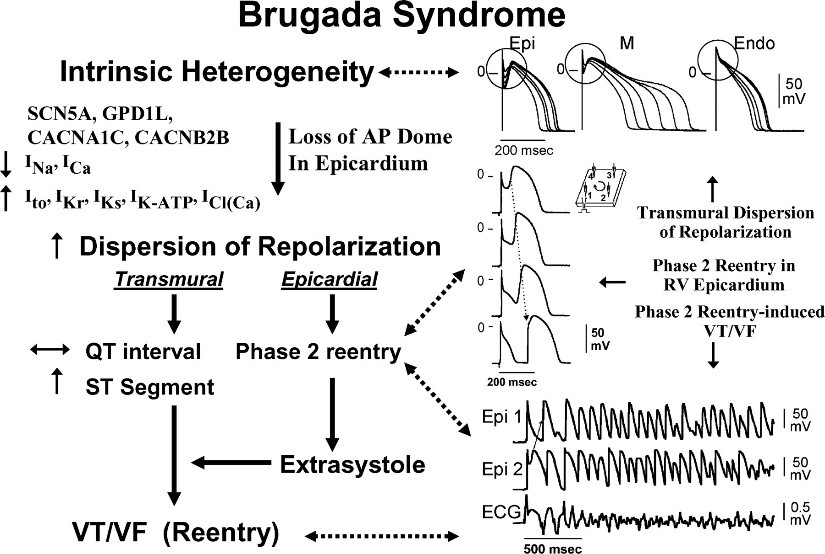
Figure 2: Schematic representation of the influence of genes involved in Brugada syndrome with the flow of ions in different phases of heart rate according to heart rate voltage [1].
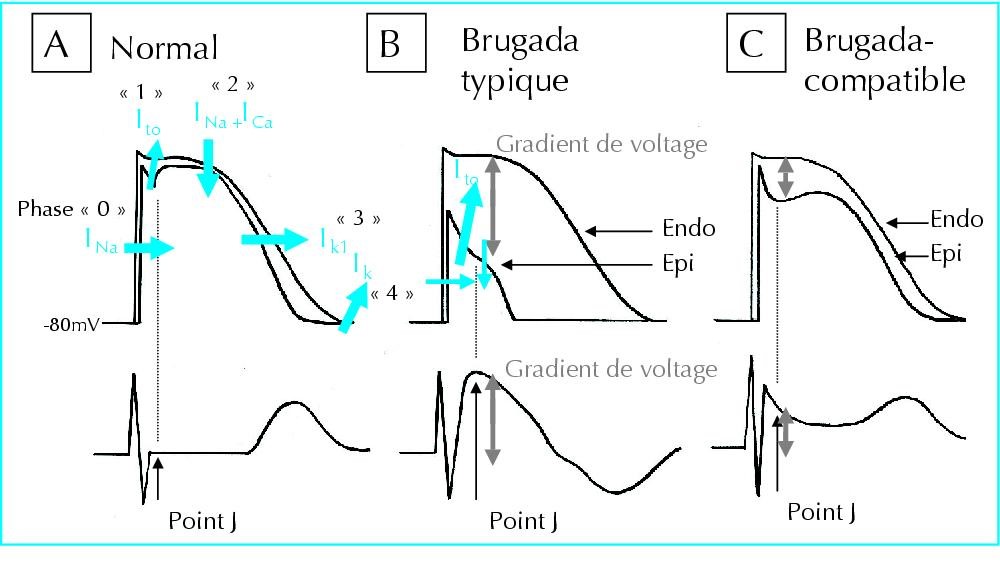
Figure 3: Schematic of the flow of sodium, calcium, and potassium ions in the normal heart rate rhythm compared to the heart with Brugada syndrome [1].

Figure 4: Schematic view of chromosome 3 where the SCN5A gene is located in the short arm of this chromosome as 3p22.2 [1].
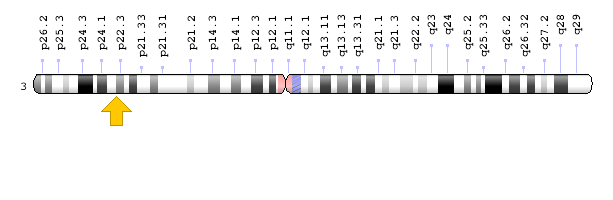
Figure 5: Schematic of chromosome 3 where the GPD1L gene is located in the short arm of this chromosome as 3p22.3 [1].
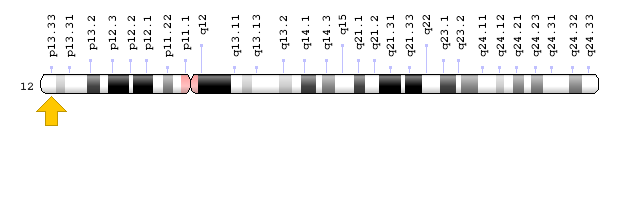
Figure 6: Schematic of chromosome 12 where the CACNA1C gene is located in the short arm of this chromosome as 12p13.33 [1].
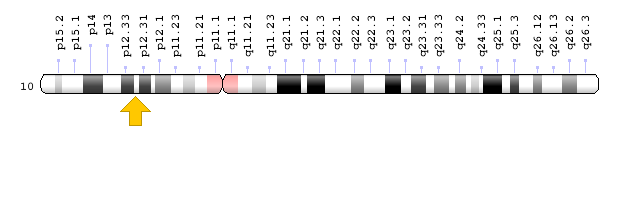
Figure 7: Schematic of chromosome 10 where the CACNB2 gene is located in the short arm of this chromosome as 10p12.31-p12.33 [1].
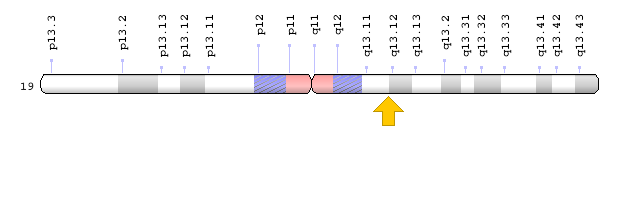
Figure 8: Schematic of chromosome 19 where the SCN1B gene is located in the long arm of this chromosome as 19q13.11 [1].
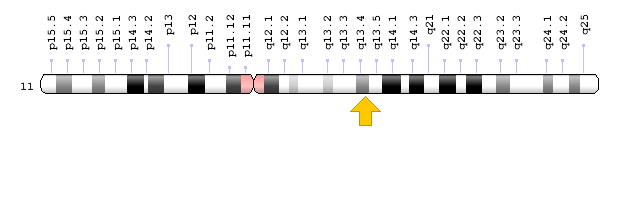
Figure 9: Schematic view of chromosome 11 where the KCNE3 gene is located in the long arm of this chromosome as 11q13.4 [1].
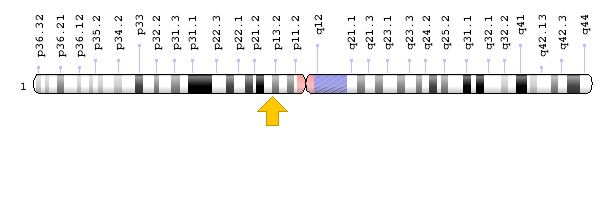
Figure 10: Schematic view of chromosome 1 where the KCND3 gene is located in the short arm of this chromosome as 1p13.2 [1].
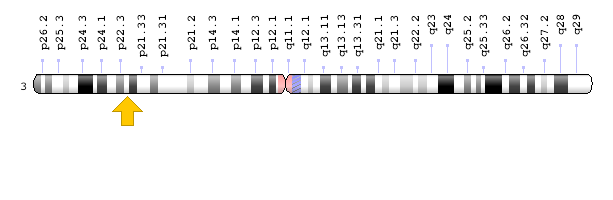
Figure 11: Schematic of chromosome 3 where the SCN10A gene is located in the short arm of this chromosome as 3p22.2 [1].
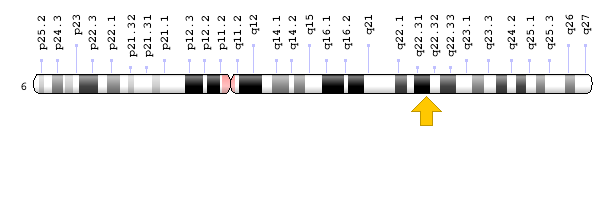
Figure 12: Schematic view of chromosome 6 where the HEY2 gene is located in the long arm of this chromosome as 6q22.31 [1].
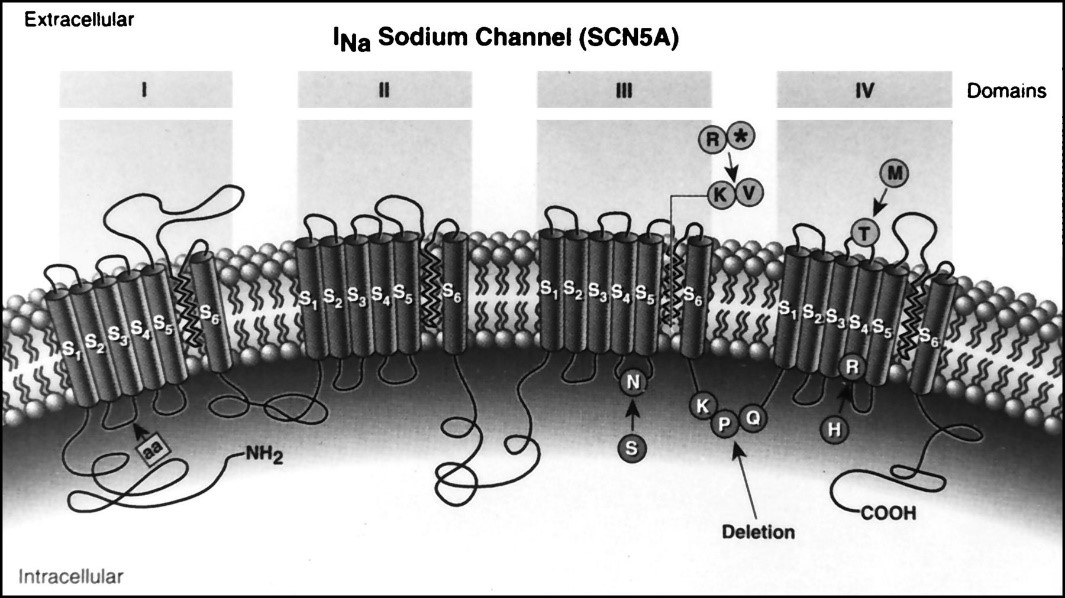
Figure 13: Schematic view of the structure and mechanism of action of the sodium channel [1].
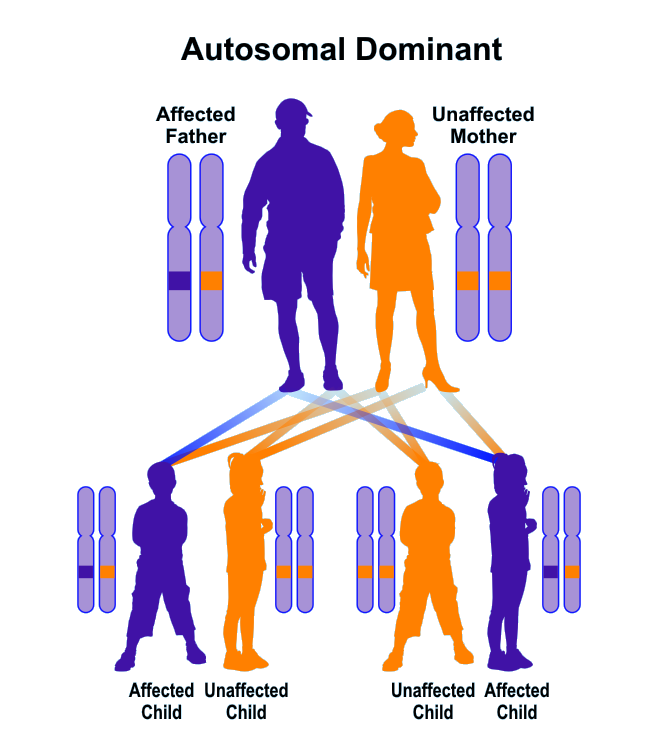
Figure 14: Schematic representation of the dominant autosomal inherited pattern that Brogada syndrome also follows [1].
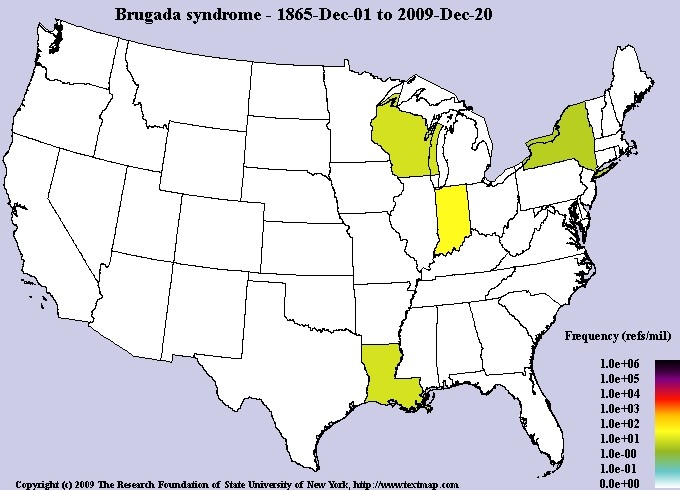
Figure 15: Schematic of the frequency map of the prevalence of Brugada syndrome in the world [1].
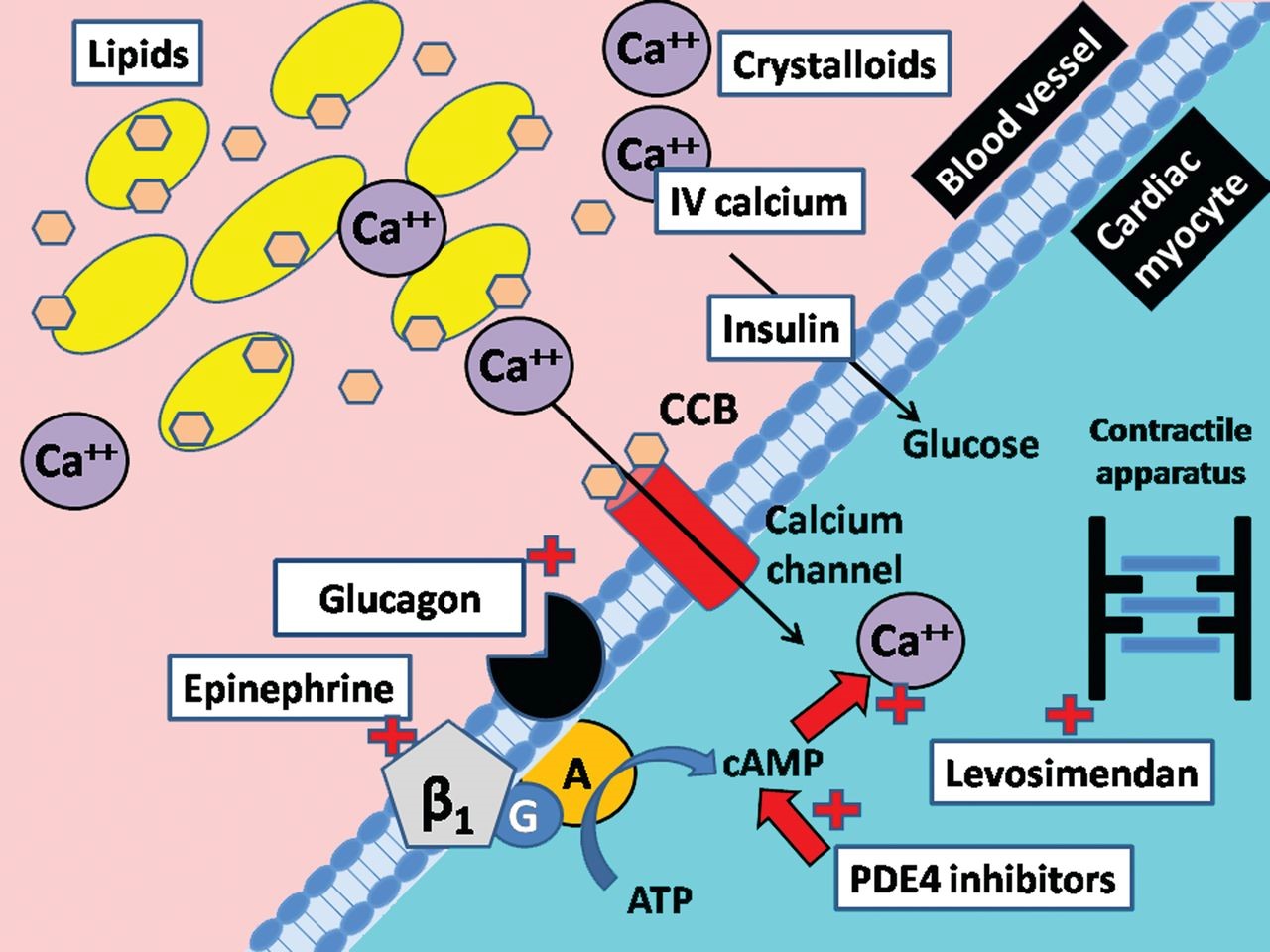
Figure 16: Schematic of calcium channel structure and function [1].
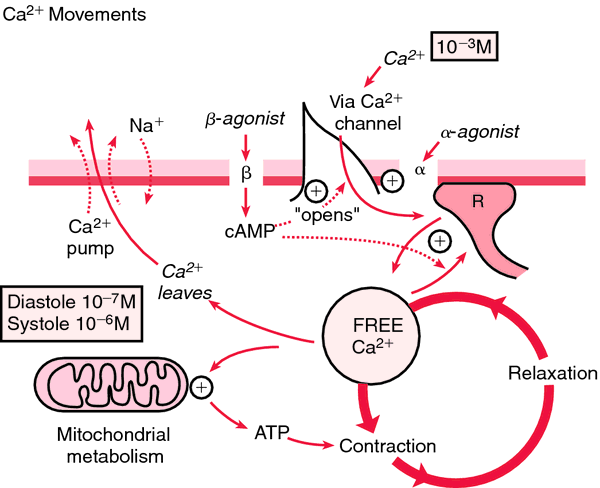
Figure 17: Schematic of how calcium is pumped in diastolic and systolic blood pressure with mitochondrial metabolism [1].
Citation: Asadi S and Yousefi R (2020)The Role of Genetic Mutations in Genes CACNA1C, CACNB2, SCN1B, KCNE3, KCND3, SCN10A, HEY2, SCN5A, GPD1L in Brugada Syndrome. Curr Trends Genet Microbiol: CTGM-100002
