
Annals of Medical & Surgical Case Reports
ISSN 2652-4414
Case Report
A Patient with Sickle Cell Disease and Beta-Mannosidosis-A Case Report
Almadani H, Namnqani R*and Kurdi H
Consultant of pediatric hematology-oncology Department of pediatrics, King Fahad Armed Forces Hospital, Jeddah, Kingdom of Saudi Arabia,
*Corresponding author: Dr. Housam Almadani , Consultant of pediatric hematology-oncology Department of pediatrics, King Fahad Armed Forces Hospital, Jeddah, Kingdom of Saudi Arabia, Tel: +966 50 468 5495; Email: dr_housam_almadani@hotmail.com
Citation: Almadani H, Namnqani R, Kurdi H (2020) A Patient with Sickle Cell Disease and Beta-Mannosidosis-A Case Report. Ann Med &Surg Case Rep: AMSCR-100061
Received date: 11 May, 2020; Accepted date: 06 June, 2020; Published date: 15 June, 2020
Abstract
Sickle Cell Disease (SCD) is one of the global health problems, with estimates of approximately 300,000 new cases per year di- agnosed with SCD worldwide. It is defined as a homozygous status of the sickle hemoglobin (HbS) gene which results in substitution of the amino acid valine to glutamic acid at the 6th position of the B-globin chain. B-mannosidosis is a rare glycoprotein lysosomal storage disease inherited as an autosomal recessive pattern caused by a deficient activity of beta manosidase enzyme. We report here an Eight years old male patient diagnosed with Sickle Cell Disease and Beta-mannosidosis, to our knowledge this is the first case to report a patient with SCD and B-mannosidosis.
Keyword: Acute Chest Syndrome; Beta mannosidosis; Hypersplenism; Sickle Cell Disease; Splenic Sequestration
Introduction
Sickle Cell Disease (SCD) is one of the global health problems, with estimates of approximately 300,000 new cases per year diagnosed with SCD worldwide. It is defined as a homozygous status of the sickle hemoglobin (HbS) gene which results in substitution of the amino acid valine to glutamic acid at the 6th position of the B-globin chain [1,2]. B-mannosidosis is a rare glycoprotein lysosomal storage disease inherited as an autosomal recessive manner caused by a deficient activity of beta manosidase enzyme [3].Acute Chest Syndrome (ACS) is the leading cause of death in SCD patient and the 2nd most common complication of Sickle Cell Disease, after vaso-occlusive crisis[4]. Ssplenic sequestration and hypersplenism is a known complication as well of SCD [5]. we report a case of SCD with B-mannosidosis who is having a recurrent ACS and hypersplenism with acute splenic sequestration.
Case report
An 8 years old boy, a product of a full term, Spontaneous Vaginal Delivery (SVD), the second childtoaconsanguineousparent. The patient presented at age of 4 months with pallor and shortness of breath with a complete blood count hemoglobin level of 7.8 mg/ dl with a sickle cell screen positive, and diagnosed as a sickle cell disease based on hemoglobin electrophoresis (HbS: 34.7% and HbF: 63.7%with no detectable HbA) which was repeated at age of one year and confirmed the diagnosis of SCD with alpha-Thalassemia trait, X-ray chest showed cardiomegaly and Echo was done and results were as dilated left ventricle and atrium with moderate mitral regurgitation and the left ventricle function of FS 17.2% and labelled as having cardiomyopathy. At age of 3 years the patient had global developmental delay, growth parameters were below 3rd centile for height and weight, he had hepatosplenomegaly, speech retardation with hearing loss and recurrent otitis media requiring later bilateral myringotomy, T tube insertion and a hearing aid. He had coarse facial features (gargoyle-like features) as frontal bossing, flat nasal bridge, large tongue with thick lips and gapped teeth, gibbus malformation with skeletal deformities (Figure 1-4) (Table 1, 2).
At 4 years of age he was diagnosed as Bronchial Asthma, Allergic Rhinitis, myopic astigmatism and Attention Deficit Hyperactive Disease (ADHD) with a lower IQ of 70 at age of 7 years.He underwent splenectomy at age of 5 years.The patient was investigated for a lysosomal storage disorder, urine Glycosamino glycan (GAG) was negative for the patient, normal blood Tandem mass spectrometry, and chromosomal analysis of 46, XY.
Whole ExomeSequence (WES) detected a homozygous variant of exon 6 of the MANBA gene mutation C.704T>G (p.lle235Arg) and both parents are with a heterozygous status. Enzymatic assay of manosidase activity in serum (Table 1).
He has a sister who had the same presentation of coarse facial features, hepatosplenomegaly, and gibbus malformation with sickle cell Trait and Beta-mannosidosis confirmed by genetic whole exome sequencing (homozygous status of MANBA gene mutation) C.704T>G) and enzyme assay (Figure5).
Discussion
Beta-mannosidosis is a pan-ethnic disorder with an autosomal recessive inheritance [6]. Two siblings from Saudi Arabia, described in our report, was associated with gargoyle like facial dysmorphism, hearing impairment, mental retardation, and recurrent respiratory tract infections.To our knowledge the number of patients reported in the literature is small, (Table 2) so, it is difficult to conclude specific symptoms and signs to characterize B-mannosidosis.Various degrees of developmental delay, hearing loss, and mental retardation are common findings. The older sibling has angiokeratoma as an isolated observation as reported previously[21,22]. The observed skeletal changes in our proband are beaked vertebral bodies, thick calvarium, bullet shaped vertebrae, and gibbus malformation (Figure.2).
This patient who is a known case of hypersplenism with multiple splenic sequestration with one intensive care admission with critical hemoglobin level 3.4 mg/dl. Post elective splenectomy the patient had significant decreased rate of admission and blood transfusions. It was noticed that in our proband who has beta- mannosidosis the most frequent complication of sickle cell disease is hypersplenism with sequestration and acute chest syndrome with fewer painful crisis’s although it is the most common feature of sickle cell disease[23]. The patient had frequent admissions as an acute chest syndrome in which after starting azithromycin as an anti-inflammatory agent[24,25] by our pulmonologist, the rate of acute chest syndrome related admission decreased.
The association of Beta mannosidosis and SCD to our knowledge has not been reported before, this combination of both diseases need further reported cases to establish the impact.
Conclusion
This is the first case to report a patient with SCD and B-mannosidosis. We recommend splenectomy for cases of Sickle Cell Disease with B-mannosidosis and hypersplenism, we recommend using azithromycin as an anti-inflammatory prophylactic agent to reduce Acute Chest Syndrome in patient with Sickle Cell Disease andBeta mannosidosis.
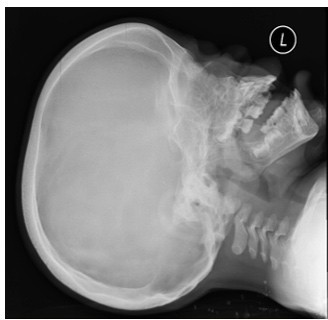
Figure 1:X-ray skull showing calvarial thickening and bullet shaped vertebrae.

Figure 2:X-ray of right upper limb showing expansion of medullary spaces with short tubular bones of the hand.
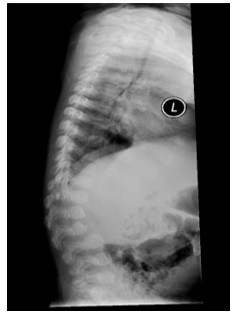
Figure 3: X-ray lateral spine showing L2 vertebral breaking with gibbus malformation.
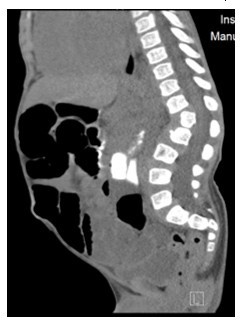
Figure 4: CT Abdomen showing anterior inferior beaking of L2 vertebrae with gibbus malformation.
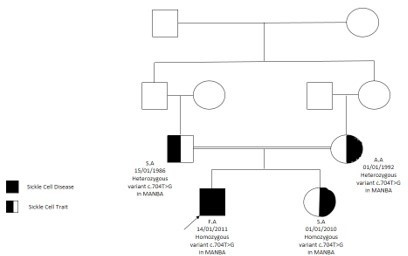
Figure 5: Family pedigree
|
Source |
enzyme |
Activity |
Reference range |
|
Serum |
Beta-mannosidase |
0.004 umol/ml/min |
1.49- 8.33 umol/ml/min |
|
Serum |
Alpha-mannosidase |
0.14 umol/ml/min |
0.1 -0.2 umol/ml/min |
Table1: Enzymatic assay of Mannosidase enzyme activity in our proband.
|
TABLE 2. Beta-mannosidosis phenotype and laboratory findings in reported cases |
|||||||||||
|
Case |
1 |
2 |
3 |
4 |
5 |
6 |
7 |
8 |
9 |
10 |
11 |
|
Sex |
Male |
Male |
Male |
Male |
Male |
Female |
Male |
Female |
Female |
Male |
Male |
|
Ethnicity |
European |
Hindu |
Hindu |
Turkish |
Turkish |
Czech |
Czech |
Jamaican |
Turkish |
European |
African |
|
Consanguinity |
Negative |
|
|
Positive |
positive |
|
|
Negative |
positive |
Negative |
Negative |
|
Diagnosis age (Yr) |
1.5 |
44 |
19 |
8 |
6 |
20 |
30 |
1 |
6 |
3 |
14 |
|
Presenting symptom |
Dysmorphology |
Mental retardation |
|
Feeding difficulty |
|
Developmental delay |
|
Seizures |
Absent speech |
Speech impairment |
Speech impairment |
|
Mental retardation |
Positive |
Positive |
Yes |
Yes |
Yes |
Positive |
Positive |
|
Positive |
Positive |
Negative |
|
Behavioral problems |
Hyperactive |
aggressive |
|
Troublesome |
|
Aggressive |
|
|
Hyperactive aggressive |
Hyperactive |
Disinterest |
|
Hearing impairment |
Mild |
Present |
Present |
Present |
Present |
Present |
Present |
Negative |
Present |
Negative |
Negative |
|
Neurological |
Speech retard |
|
|
Speech retard |
|
Developmental delay |
|
Developmental delay |
Motor delay |
Speech impairment |
Peripheral neuropathy |
|
dermatologic |
|
Angiokeratoma |
|
|
|
Erysipelas |
|
|
|
|
|
|
Facial dysmorphism |
Present |
negative |
Negative |
Negative |
Negative |
Present |
Present |
Present |
Present |
Present |
Negative |
|
Skeletal deformation |
Present |
Negative |
Negative |
Negative |
Negative |
Present |
Present |
Negative |
Present |
Negative |
Negative |
|
Respiratory infections |
|
|
|
Positive |
positive |
Positive |
Positive |
|
Positive |
Positive |
Negative |
|
mannosidase |
Deficient |
Deficient |
Deficient |
Deficient |
Deficient |
Deficient |
Deficient |
Deficient |
Deficient |
Deficient |
Deficient |
|
Urine disaccharides |
Positive |
Positive |
Positive |
Positive |
positive |
Positive |
Positive |
Positive |
Positive |
Positive |
Faint |
|
Reference |
Wenger et al. 1986[7] |
Cooper et al. 1986-1988[21,22]] |
Cooper et al. 1986-1988[21,22] |
Dorland et al. 1988[8] |
Dorland et al. 1988[8] |
Kleijer et al. 1990[6] |
Kleijer et al. 1990[6] |
Cooper et al. 1991[9] |
Wijburg et al. 1992[10] |
Poenaru et al. 1992[11] |
Levade et al. 1994[3] |
|
Cont. TABLE 2. Beta-mannosidosis phenotype and laboratory findings in reported cases (To our knowledge) |
Sibling sister |
our case |
||||||||||
|
Case |
12 |
13 |
14 |
15 |
16 |
17 |
18 |
19 |
20 |
21 |
22 |
23 |
|
Sex |
Female |
Female |
Female |
Female |
Male |
Male |
Female |
Female |
Male |
Female |
female |
Male |
|
Ethnicity |
white |
|
Arabic |
Arabic |
Japanese |
French |
Spanish |
Arabic |
Algerian |
Asian |
Arabic |
Arabic |
|
Consanguinity |
Negative |
Positive |
Positive |
positive |
Positive |
negative |
|
Positive |
Positive |
Positive |
positive |
Positive |
|
Diagnosis age |
22 years |
7 months |
2 years |
3.5 years |
51 years |
18 years |
24 years |
36 years |
12 years |
5 months |
7 years |
5 years |
|
Presenting symptom |
clumsiness |
Feeding difficulty |
Seizures |
Seizures |
Angiokeratoma |
Developmental delay |
Angiokeratoma |
Angiokeratoma |
Clumsiness |
Seizures |
Dysmorphism |
Dysmorphism |
|
Mental retardation |
Negative |
|
Positive |
positive |
Positive |
Positive |
Negative |
Positive |
Positive |
Positive |
Positive |
Positive |
|
Behavioral problems |
Scantly communicative |
|
|
|
|
ADHD |
Negative |
Aggressive |
|
|
ADHD |
ADHD |
|
Hearing impairment |
Negative |
|
Negative |
negative |
Present |
Present |
Mild |
Present |
Present |
Present |
Present |
Present |
|
Neurological |
Negative |
Developmental delay |
Developmental delay, speech delay |
encephalopathy |
Peripheral neuropathy |
Developmental delay, speech delay |
Negative |
|
Cerebellar ataxia |
Developmental delay |
Developmental delay |
Developmental delay |
|
dermatologic |
Angiokeratoma |
|
Negative |
Negative |
Angiokeratoma |
Negative |
Angiokeratoma |
Angiokeratoma |
Negative |
|
Angiokeratoma |
Negative |
|
Facial dysmorphism |
Negative |
Negative |
Negative |
Negative |
Negative |
Negative |
Negative |
Negative |
Negative |
Positive |
Positive |
Positive |
|
Skeletal deformation |
Negative |
Negative |
Negative |
Negative |
Negative |
Negative |
Negative |
Negative |
Negative |
Positive |
Positive |
Positive |
|
Respiratory infections |
Negative |
positive |
Negative |
Negative |
|
Positive |
|
Negative |
|
Positive |
Positive |
Positive |
|
mannosidase |
Deficient |
Deficient |
Deficient |
Deficient |
Deficient |
Deficient |
Deficient |
Deficient |
Deficient |
Deficient |
Deficient |
Deficient |
|
Urine disaccharide |
Positive |
Positive |
|
|
|
Positive |
Positive |
|
|
|
Positive |
Positive |
|
Reference |
Rodriguez et al. 1996[12] |
Gourrier et al. 1997[13] |
Cherian et al. 2003[14] |
Cherian et al. 2003[14] |
Suzuki et al. 2004[15] |
Sedel et al. 2006 [16] |
Gort et al. 2006 [17] |
Molho et al. 2007 [18] |
Levade et al. 2009[19] |
Broomfield et al. 2012[20] |
Almadani et al 2019 |
Almadani et al. 2019 |
Citation: Almadani H, Namnqani R, Kurdi H (2020) A Patient with Sickle Cell Disease and Beta-Mannosidosis-A Case Report. Ann Med &Surg Case Rep: AMSCR-100061
