
Annals of Medical & Surgical Case Reports
(ISSN 2652-4414)
Case Report
Case Reports of Congenital Pyloric Atresia Laparoscopic Repair
Abbas W*, Lam J and Hammond P
Department of Paediatric Surgery, Royal Hospital for Sick Children Edinburgh, UK
*Corresponding author: Wisam Abbas,Department of Paediatric Surgery, Royal Hospital for Sick Children Edinburgh, UK.
Citation: Abbas W, Lam J, Hammond P (2020) Case Reports of Congenital Pyloric Atresia Laparoscopic Repair. Ann Med &Surg Case Rep: AMSCR-100049
Received date: 18 March, 2020; Accepted date: 20 March, 2020; Published date: 27 March, 2020
Abstract
This paper reports two cases of isolated congenital pyloric atresia (CPA) in neonates repaired using laparoscopic approach. We believe this is the second article in published literature describing minimal invasive repair of this rare condition. Both patients had uneventful post-operative course and were discharged on full feeding with satisfactory outpatient follow up.
Keywords: Congenital pyloric atresia; Gastrointestinal atresia; Neonates
Introduction
Congenital pyloric atresia (CPA) is a rare condition. It accounts for less than 1% of all gastrointestinal atresia with a reported incidence of 1:100,000 live births [1]. CPA may occur as an isolated lesion and in about half of the cases it is associated with other congenital anomalies such as epidermolysis bullosa (EB), aplasia cutis congenital (ACC), intestinal atresia and oesophageal atresia [2]. Associated anomalies are a contributing factor for the reported high mortality [1]. Laparotomy and surgical repair is well established surgical treatment. We present our experience in laparoscopic management of two cases of isolated CPA. We believe this is the second report after one case published from Vietnam by SON AND HOAN [2].
Patient 1: Baby boy with no antenatal polyhydramnios was born at term with a birth weight of 3.7 kg. He wast ransferred to our centre on day three of life with dehydration, non-bilious vomiting andweight loss. He passed meconium on day one of life.At presentation he was haemodynamically stable, abdomen soft and non-distended with no associated skin abnormality. Abdominal film showed distended stomach with gastric peristaltic wave. Scanty gas noted in distal bowel(Figure 1). Upper GI contrast performed to rule out malrotation with midgut volvulus.
Laparoscopy performed the following day after stabilisation. Body weight at the time of surgery was 3.3 kg. Laparoscopic approach through umbilical 5-mm camera port and 3mm working ports at the right and left lower quadrants. Laparoscopic view showed dilated stomach and Liver was elevated by hitching falciform ligament up to anterior abdominal wall with a percutaneously inserted 4/0 polydioxanone suture. There was no obvious calibre change between the proximal and distal parts of the duodenum. A longitudinal duodenotomy was made in the first part of the duodenum and bile stained fluid was noted excluding the diagnosis of a preampullary duodenal web. 5 French feeding tube was inserted via the duodenotomy and advanced distally down the duodenum but was met with resistance proximally towards the stomach. The pyloric wall was thickened and the enterotomy therefore extended proximally through the pylorus with findings consistent with thickened pyloric mucosal membrane. The enterotomy was then closed transversely in the form of A Heineke-Mickulicz pyloroplasty using interrupted 5/0 Polyglactin.6 French Trans-anastomotic tube inserted.
The patient was admitted to surgical ward after operation. Naso-jejunal feeding was started at postoperative day one and progressed to full feed after 7 days. Trans-anastomotic tube removed on day 11 after full NG feeding established. Patient was on full oral feed two weeks after the surgery and discharged on day 15.
Outpatient review at 5 months of age showed the child was feeding well and started weaning to solids and gaining weight (7.9kg above the 50th centile).
Patient 2: A boy with antenatal polyhydramnios was born at term with a birth weight of 2.8 kg. He was transferred to our hospital on day three with failure to establish feeding and non-bilious vomiting. He had passed meconium on day one of life. At presentation he was haemodynamically stable, abdomen soft and non-distended with no associated skin abnormality. Capillary blood gas was normal. Abdominal radiograph demonstrated a dilated single gastric bubble with no distal bowel gas (Figure 4).
Abdominal ultrasound confirmed the distended stomach with the pylorus being in serosal continuity with an empty duodenal lumen suggestive of pyloric obstruction. The orientation of the superior mesenteric artery & vein was normal with no evidence of malrotation/volvulus.
Following stabilization laparoscopy was performed the following day (with a 5mm umbilical port and 3mm ports at the right & left flanks and epigastrium). The duodeno-jejunal flexure was confirmed to the left of midline. There was serosal continuity of the pylorus and first part of the duodenum. The site of obstruction with calibre change was identified following inflation of the stomach via the nasogastric tube. A longitudinal incision of the pylorus extending 1.5 cm proximal and distal to the apparent obstruction site was performed.
The nasogastric tube was identified proximally and the tip passed distally into the second part of the duodenum before being withdrawn. A pyloric membrane was excised and the pyloroplasty was subsequently closed transversely with interrupted 5-0 polyglactin (Vicryl) suture. There was no evidence of leakage on inflation of the stomach and the omentum was laid over the anastomosis.The patient was extubated the following day. Water soluble contrast on day 4 showed passage of contrast into the duodenum with no leak (Figure 5) with subsequent initiation of oral feeds increasing slowly to full feeds after 12 days and discharge a week later at 3.2 kg body weight. Clinical review at eight months of age showed that he was thriving had weaned to solids with weight of 8.7 kg (between 25thand 50thcentile).
Discussion
The first case of CPA was described by Calder in 1749. Touroff and colleagues performed the first successful operation in 1940[4]. CPA is known to be associated with low birth weight and polyhydramnios, both of which are reported in up to 60% of the patients. Overall mortality is very high, exceeding 50%, but this is due to the high incidence of severe and often fatal associated anomalies [3]. CPA can be divided into three clinical types: Isolated, associated with other genetically determined conditions namely EB and/or ACC and CPA associated with other gastrointestinal atresia which are usually multiple [1].
The exact aetiology of CPA is unknown. It may result from failure of the pyloric tube to canalise during embryonic development. Other theory proposed a mechanical cause or vascular accident. CPA associated with epidermolysis bullosa (EB) results from Inflammatory responses lead to development of secondary fibrosis leading specifically to the obstruction of the intestinal lumen especially in anatomically narrow spaces, such as pylorus [5].
Anatomically, there are 3 recognized varieties of CPA. Type 1: pyloric membrane or diaphragm. This is the most common type reported in 60% of patients. Type 2: pyloric atresia with no gap. Type 3: pyloric atresia with a gap [1].
The first patient had normal antenatal scans whereas the second case did have polyhydramnios. Both patients presented with non-bilious vomiting, failure to establish feeds and weight loss. Both of our cases were isolated CPA. First patient had abdominal radiograph which showed dilated stomach with paucity of distal gas followed by upper GI contrast study which ruled out malrotation with midgut volvulus and was consistent with gastric outlet obstruction. Second patient had abdominal radiograph which showed dilated stomach with no distal bowel gas.
The laparoscopic approach was suitable in both patients after consideration of several factors including. Gestational age, body weight, isolated anomaly, Instrument availability and paediatric surgical & Anaesthetic expertise.According to our literature search, there has been only one case report of laparoscopic repair of CPA from Vietnam by SON AND HOAN [2]. We think it is difficult at this stage to draw statistical conclusions in comparing laparoscopic and open surgical repair until availability of further reports to constitute statistically significant sample size. We understand the rarity of the condition which contributes to slow building up of case numbers and accumulation of experience.
Conclusion
Congenital pyloric atresia is a very rare surgical condition. Two case of isolated CPA repaired using laparoscopy reported in this paper. We think minimal invasive approache repair is feasible and can be benefical in reducing morbidity related to laparotomy and may paly a role in decreasing time required to esablish full feeding. We acknowledge that this report does not constitute a strong evidence and further studies required to prove the statistical significance of laparoscopic approach.
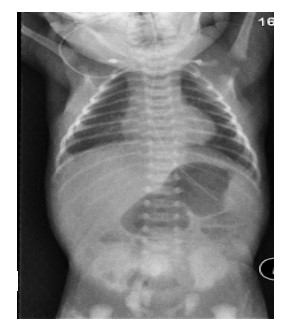
Figure 1:Abdominal film showed distended stomach with gastric peristaltic wave. Scanty gas noted in distal bowel
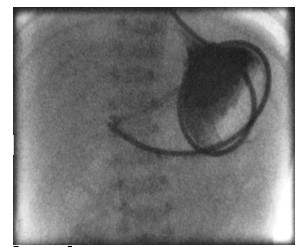
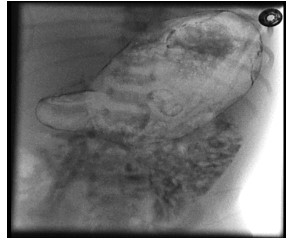
Figure 2, 3:There was no progression of contrast beyond the gastric outlet despite multiple attempts by radiologist to pass the tube through the pylorus(Figure 2,3).
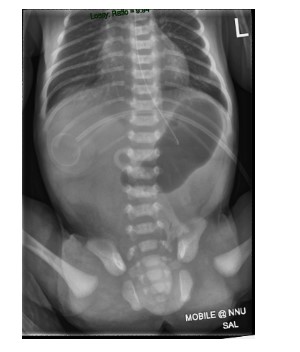
Figure 4: Abdominal radiograph demonstrated a dilated single gastric bubble with no distal bowel gas
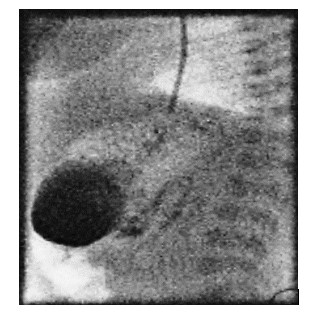
Figure 5: Water soluble contrast on day 4 showed passage of contrast into the duodenum with no leak.
Citation: Abbas W, Lam J, Hammond P (2020) Case Reports of Congenital Pyloric Atresia Laparoscopic Repair. Ann Med &Surg Case Rep: AMSCR-100049
