
Journal of Surgery and Insights
(ISSN 2652-4643)
Case Report
Atypical Presentation of Cystic Fibrosis with Cyanotic Congenital Heart Disease
Incheti G, Taksande AM*, Meshram R and Lohakare A
Department of Paediatrics, Jawaharlal Nehru Medical College, India
*Corresponding author: Amar .M. Taksande, Department of Paediatrics, Jawaharlal Nehru Medical College, India, E mail: amar.taksande@gmail.com
Citation: Incheti G, Taksande A, Meshram R and Lohakare A (2020) Atypical presentation of cystic fibrosis with cyanotic congenital heart disease. J Surg Insights: JSI-100018
Received date: 20 March, 2020; Accepted date: 23 March, 2020; Published date: 30 March, 2020
Abstract
Cystic fibrosis (CF) is the most common genetically inherited condition in western populations. The presentation of CF includes chronic respiratory infections and malabsorption. Here, we report the CF case diagnosed in infancy with cyanotic congenital heart disease.
Keywords: Congenital; Cyanotic; Cystic Fibrosis; Heart disease
Introduction
Cystic fibrosis (CF) is an inherited as an autosomal recessive trait with multisystem disorder of children. The Cystic Fibrosis Trans-membrane Conductance Regulator (CFTR) gene located at 7q 31.2, which regulates the activity of chloride and sodium channels at the surface of the epithelium cell [1-3]. This disease affects the cells that produce mucus and sweat in multiple organs, being the lung the most severely affected and responsible for 90% of the deaths in patients with CF. The dysfunction of the CFTR protein, leads to a wide and variable array of presenting manifestations and complications. Although the respiratory and gastro-intestinal symptoms are well recognised, the cardiac involvement of CF is uncommon [2-4]. In this case report, we describe the CF case diagnosed in infancy with cyanotic congenital heart disease (CHD).
Case Report
A 9 month old male child born out of 3rd degree consanguinity, admitted with complaints of fever, cough and difficulty in breathing. He was delivered via normal vaginal delivery with birth weight of 2.5 kg and had history of NICU stay in view of congenital pneumonia and also developed myoclonic type of convulsion. However, he was discharge in stable condition without complication. He had 4 normal female siblings and 2 male siblings who were died with similar complaints at the age of 2 months, as 1st one had pneumonia and the second with congenital heart disease (Figure 1). Patient had multiple hospital admissions with similar complaints.
His weight was 6 kg, height of 64cm and head circumference was 39 cm. According to WHO, child was moderately wasted and severe stunted. His vitals were: HR: 118/min, RR: 52/min, BP: 84/56 mmHg and Spo2: 88%. On general examination, patient had minimal central cyanosis with no clubbing. Signs of respiratory distress were present with nasal flaring, tachypnoea and subcostal retractions. On respiratory examination, bilateral crepitations were present. On cardiovascular examination, the ejection systolic murmur was present, best heard at 2nd left parasternal area. Others systemic examination was normal.
The haematological work up was normal. C-reactive protein was elevated. Chest x-ray showed normal size heart with oligemic lung field. Electroencephalogram was suggestive of multifocal epileptiform abnormalities. MRI brain showed hyperintensities in perirolandic region. Blood culture was suggestive of growth of pseudomonas species sensitive to ciprofloxacin and cotrimaxozole. Echocardiography study demonstrated Tetralogy of Fallot (Large size, bidirectional, Ventricular septal defect with Moderate valvular pulmonary stensosis). Urinary examination showed elevated tyrosine metabolites. Whole exome gene sequencing was done with EDTA whole blood which was suggestive of cystic fibrosis. Patient was given oxygen support, intravenous fluids, and antibiotics and continued with antiepileptics. As patient had lower respiratory tract infection with multiple fever spikes, antibiotics were upgraded accordingly. Nevertheless, his condition continued to deteriorate. He became increasingly cyanotic and dyspenic. After about 20 days of hospital stay, patient required inotropic support as he was unable to maintain saturation with poor perfusion. Later, patient was died inspite of giving best treatment.
Discussion
Cystic fibrosis (CF) is an inherited multisystem disorder of children. The earliest symptom is usually cough that may begin with a viral respiratory tract infection but then persists unless treated with antibiotics. Child may present with typical CF-related symptoms such as chronic respiratory infections or malabsorption. Patients with atypical disease tend to present late in childhood or as adults with less widely known complications such as pancreatitis, congenital absence of the vas deferens and azoospermia or nasal polyps. Cor pulmonale, respiratory failure, and death eventually supervene unless lung transplantation is accomplished. Following the lungs, the GIT are severely affected since the mucous plugs obstruct pancreas enzymes and bile flow into the duodenum resulting in digestion abnormalities and malabsorption [1-4]. In 15-20% of newborn infants with CF, the ileum is completely obstructed by meconium (meconium ileus).
It’s characterized by the classic triad of elevated sweat chloride levels, chronic sinopulmonary disease, and pancreatic insufficiency. The diagnosis in the first year of life is typically accompanied of meconium ileus, failure to thrive, pulmonary infections, diarrhea, and steatorrhea. Severe, untreated pancreatic insufficiency can present with protein energy malnutrition, anasarca, hypoproteinemia, electrolyte loss, anemia, and failure to thrive. Nutritional deficiency dermatitis may be present as the initial manifestation of CF [3-6]. Acute cardiac failure in pathologic myocardial fibrosis often characterizes cardiac involvement of cystic fibrosis in infants. Cor pulmonale as a result of chronic hypoxemia with resultant effect on the pulmonary vasculature is the dominant and a more serious presentation of cardiac involvement in older affected individuals. However, there is very little documentation regarding congenital cardiac anomalies in patients with cystic fibrosis and their effect in the prognosis of these patients [7] have reported a single case of twins affected by cystic fibrosis with concomitant cardiac anomalies; one sibling was affected by an atrial septal defect, which was treated surgically, while the other sibling was affected by a ventricular septal defect which closed spontaneously. Farooqui et al [8] reported bicuspid aortic valve with mild to moderate aortic stenosis, dilated ascending aorta, dilated superior vena cava, mild left ventricular hypertrophy, and a moderate restrictive patent ductus arteriosus with left to right shunt (PDA). In our case, there was a large size, bidirectional, Ventricular septal defect with Moderate valvular pulmonary stensosis with overriding of aorta suggestive of tetralogy of fallot.
Conclusion
The spectrum of presentation of CF is wide and varied, and this case highlights an atypical presentation CF with of tetralogy of fallot.
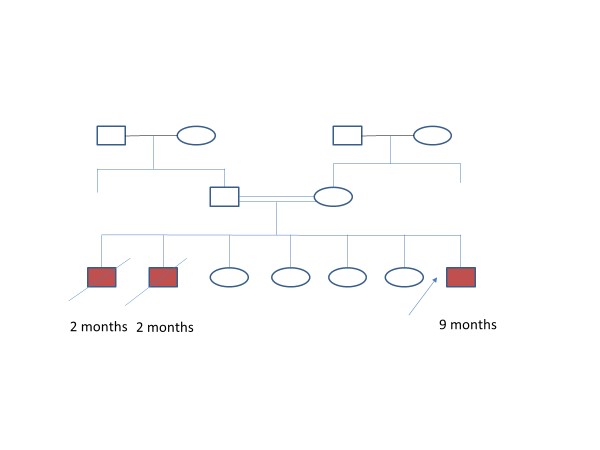
Figure 1: Pedigree chart.
Citation: Incheti G, Taksande A, Meshram R and Lohakare A (2020) Atypical presentation of cystic fibrosis with cyanotic congenital heart disease. J Surg Insights: JSI-100018
