
Current Trends in Biotechnology and Biochemistry
Research Article
Insilico Analysis and Homology Modeling of TypA (Rv1165) Protein of Mycobacterium Tuberculosis H37Rv
Meena S, Bhanawat S and Meena LS*
CSIR-Institute of Genomics and Integrative Biology, India
*Corresponding author: Laxman S. Meena, CSIR-Institute of Genomics and Integrative Biology, India, Tel: 0091-11-27002200; Email: meena@igib.res.in
Citation: Meena S, Bhanawat S and Meena LS (2019) Insilico Analysis and Homology Modeling of TypA (Rv1165) Protein of Mycobacterium Tuberculosis H37Rv. Curr Trends Biotechnol Biochem: CTBB-100008.
Received date: 29 October 2019; Accepted date: 05November, 2019; Published date: 12 November, 2019
Abstract
Tuberculosis is becoming the prodigious reason for mortality in the developing world.Thus, there is an urgent need for escalating elixir against tuberculosis.Despite the availability of Bacilli Calmette Guerin (BCG) and other vaccines against the malady, tuberculosis is a pivotal reason for millions of deaths every year because of the emergence of Multi-Drug Resistant TB (MDR-TB), Extensively-Drug Resistant (XDR-TB).Mycobacterium tuberculosis is the causative agent of tuberculosis which is a gram positive, aerobic bacterium.Tuberculosis bacteria penetrate the body through the breathing of transmittable droplet nuclei exposed to the air as droplet nuclei from sneezing, coughing of people with pulmonary or lateral tuberculosis (TB) which ultimately reaches the lungs alveolar macrophages for its duplication, causing primary infection and secondary infection in lymphoid organs.To discontinue the transmission there is a need to understand the role of each protein present in the genome of M. tuberculosis.Thus this manuscript covers some insilico approaches against non-essential, probable GTP-binding translation elongation factor typA (Rv 1165) of M. tuberculosis.TypA gene acts as a virulence factor in other organisms but in M. tuberculosis its role is in cellular processes.GTP binding protein motif DXXG is present in various positions.Insilico approaches include interaction studies with proteins and also with the ligands, model building, validation of the build model, and mutational analysis of the gene.Mutational analysis shows that protein destabilizes when at 371 positions isoleucine is replaced by threonine.Hence this study might be an important step in the way of eradication of this disease.
Keywords: Ligand binding prediction; Mycobacterium tuberculosis H37Rv; Multiple sequence alignment; Mutation analysis
Abbreviations
BCG : Bacilli Calmette Guerin
GTPases : Guanosine Triphosphatases
G-Proteins : GTP-Binding proteins
I-TASSER : Iterative Threading ASSEmbly Refinement
LOMETS : Local Meta Threading Server
M. tuberculosis : Mycobacterium tuberculosis
M. canetti : Mycobacterium canetti
M. bovis : Mycobacterium bovis
MDR-TB : Multidrug-Resistant TB
NCBI : National Center for Biotechnology Information
PDB : Protein Data Bank
RAMPAGE : Ramachandran Plot Analysis
STRING : Search Tool for the Retrieval of Interacting Genes/Protein
TB : Tuberculosis
UniProt-GOA : Gene Ontology Annotation
XDR-TB : Extremely Drug Resistant TB
XDR-TB : tyrosine phosphorylated protein A
Introduction
Tuberculosis (TB) is the preeminent reason transience and morbidity around the globe.TB is the deadliest disease which is said to affect one-third of the human population in the emerging world [1]. The factor responsible for proliferation of tuberculosis is human-to-human transmission of the ailment by aerosol route, capability of the bacterium to recur in host immune system and absenRteeism of capable vaccine against preclusion of transmission of the infection [2] and provoke tissue-damaging response [3]. Emergence of Multi-Drug Resistance (MDR-TB), Extensively-Drug Resistance (XDR-TB) and co-infection with HIV and diabetes type 2 are the fueling TB epidemics [4]. According to global epidemiological studies, WHO states that 10.0 deaths in 2017 which included 5.8 million men, 3.2 million women and 1.0 million children [5]. Etiological agent for tuberculosis is M. tuberculosiswhich has been evolved from M. bovis that is the agent of bovine tuberculosis [6]. Robert Koch was the first one to isolate and analyze the bacterium causing TB [7]. M. tuberculosisis the member of MTBC complex which includes other 6 other bacteria such as M.canetti, M. bovis and others [8]. Argumentation trailling for the chronic nature of the disease is due to key features of M. tuberculosis such as slow growth, cell cover of the bacterium and intracellular pathogenesis [9]. Transmission of the disease occurs through coughing, sneezing of the infected person from pulmonary or extra pulmonary tuberculosis [10].After transmission from infected person by the means of air droplet, the bacterium crosses the respiratory tract and enduringly resides in alveolar macrophages for a long time without interference of host immune system [11]. The potential of M. tuberculosis fully depends upon the ability of the bacterium to utilize host macrophages for replication and prominently the macrophages ought to persist in viable condition [12]. Tuberculli bacilli are versatile because of properties that can cause both latent and active ailment. M.tuberculosis can be latent for many years persisting in macrophages within granuloma before being active infection [13]. Infection is reactivated underneath immunosuppressive and other deliberating circumstance [14]. To systematize the transmission of the bacterium there is rising necessitate determining the host barrier scaffold and adding to its annihilation.According to earlier study it has been proved that GTP binding genes help in endurance of numerous bacteria.GTP-binding proteins are extensively allocated as molecular switches that usually cycle between GDP-bound “off” and GTP-bound “on” state [15]. GTP binding genes principally functions in cell signaling. Additionally, functions in translation, protein translocation, cell division and metabolic pathways [16]. GTP-binding protein interacts with downstream effectors when bound to GTP.As GTPase are conserved and important cellular processes they can be evaluated as feasible drug objective [17]. Being highly conserved and functions by ribosome binding. G1, G2, G3 and G4 are accountable for particular interaction with the guanine nucleotide and effecter molecule [18]. GTPase account for 70% of the current drug target and can be further evaluated for effective designs of experiment for formulation on anti-tuberculosis drugs [19]. Rv1165 (typA) contains concord sequence such as DXXG, GXXXXGK motif such is a probable GTP-binding protein.Formerly typA was found in Salmonella typhimurium (S. typhimurium) as a gene that induced by cationic hosts defense protein [20]. TypA is also attenuated as bipA.Open reading frame of typA of M. tuberculosisshares 48.7% sequence identity with BipA of Escherichia coli (E. coli) [21]. TypA belongs to super family of ribosome binding GTPase within the class TRAFAC class (translation factors) of GTPase [22]. These classes of GTPase are those proteins in which the activity of GTPase is induced by large ribosomal subunit [23]. In prior studies it has been stated that typA is a global virulence factor and help in stress adaptation in other bacteria such as E. coli and Salmonella [24].TypA helps in regulation of cell processes that constitute of virulence, symbiosis resistance to defense mechanism of host, protein trafficking by attachment to new polypeptide that has been synthesized [25] and defense against antibiotics [26]. Therefore, in this literature, we have elucidated some bioinformatics approaches against about GTP binding protein known as Rv1165 (typA) of M. tuberculosis. The study consists of assessment of typA of M. tuberculosis with other bacteria having TypA gene, search various interactive proteins and mutation the gene at GTP binding motif ought to alter the stability of the protein.Thus these studies might augment experimental procedures in enhancement of anti-tuberculosis drug.
Material and Methods
Multiple Sequence Alignment
To align more than two sequences that may be a protein or a nucleotide of similar length multiple sequence alignment is used.Certain predictions ended by means of multiple sequence alignment are evolutionary analysis, conserved domains, and to find diagnostic pattern to characterize protein family [27]. Clustal omega is used for alignment of sequences.Clustal omega uses a hidden Markov model (HMM) which is an alignment engine [28]. Clustal Omega can be accessed bymedium-large sized sequence can be processed in clustal omega [29]. Rv1165 of M. tuberculosisis aligned with other bacteria such as Escherichia coli K-12, Azospirillum brasilense, Moraxella catarrhalisBBH18, Shigella flexneri, Pseudomonas aeruginosa that have the same gene.
Protein-Protein & Protein-Compound Interaction
STRING (Search Tool for the Retrieval of Interacting Genes/Protein) is a global precompiled database for analysis of functional correlation between proteins.STRING illustrates both direct physical binding and that protein that interrelate indirectly by sharing a substrate in metabolic pathway or by contributing in large multi-protein assembly.Database consists of a graphical representation of interaction which presents a clear view of linkage and a unique scoring framework based on Benchmarks [30]. Predication done by STRING to compute correspondence between proteins is based upon some known experimental interactions from primary database, automated text-mining [31]. Cut off score ranges from low, medium, and high.Low score is attenuated as less than 0.4 medium is in range of 0.4 to 0.7 and more than 0.7 is classified as high [32, 33].
SWITCH is a search tool for information of chemicals creates a network by plot of interaction of specific protein with its ligands from Pubchem by merging stereo isomers and salt forms of the same molecule into the compound [34]. STITCH accrues information about metabolic pathways, crystal structures, binding experiments and drug-target relationships.The database is accessible at [35]. Interactions are derived from 5 sources such as genomic context predictions, high- throughput lab experiments, co- expression, and automated text mining and previous knowledge in databases.
Ab Initio Modelling of Rv1165
Generation of 3-D structure of protein from amino acid sequence is completed through I-TASSER (iterative threading assembly refinement).It can be found online at structure generated from I-TASSER is analyzed by the benchmark scoring system and outcomes engendered from I-TASSER include five full length models, C-score, TM score and the deviation from the estimated [36]. Formation of 3D structure is completed in four steps which include threading, structural assembly, model selection and refinement, structure-based functional annotation.For secondary structure of the protein, the tool, secretly pioneer Local Meta Threading Server (LOMETS) which uses H, E and C coherent for alpha-helix, beta-sheet and curl respectively.Accuracy of the structure is predicted according to the C-score.C-score ranges of -5 to 2, at which high value signifies higher possibilities of the predicted structure to be accurate and vice versa [37].
Validation of Predicited Model of Rv1165
The models designed are validated using SAVES Meta server.The server scrutinizes the overall structural geometry of the protein and residue-by-residue geometry which helps to check stereochemical value of the protein.For validation of the structure in course and following, model refinement this Meta server has 6 programs which include ERRAT, verify 3D, PROVE, ProSA, ProQ and RAMPAGE [38]. RAMPAGE is based on examination of Ramachandran plot. Ramachandran plot depicts the stereochemical nature of the protein by showing the favored and unfavored regions.The structure is permitted on the basis of assessment of the protein structure on the premise of ????, ????point of individual deposits [39,40].
Ligand Binding Site Analysis
Prediction of ligand binding pocket is the most imperative step for functional annotation.It is useful for structure based drug design.COACH is used for prediction of ligand binding pockets. COACH can be accessible from COACH is a Meta server which utilizes the target a file in PDB format.Ligands sites are predicted by two systems TM-SITE and S-site ligand restriction sites are predicted by the two.C-score obtained from COACH is resultant from 5 conclusions which include S-SITE, TM-SITE, FINDSITE, BS-SCORE C-SCORE and concavity.Its outcomes also include linear-SVM algorithm for combine weight assessment [41]. Coverage of global structure is denoted by cov, C-score is the confidence score and if BS-score is less than 1 indicates proportion of environs closeness among template binding site and envisage site.Cut off score, BS-score less than one reveal a noteworthy local match among the predicting and template binding site [42,43]. C-score ranges from 0-1, where higher score indicates reliable prediction.
Functional Prediction of Rv1165
Function of a protein is forecasted by insilico approaches.Server used for function prediction of target gene is COFACTOR.COFACTOR is available at COFACTOR is a structure based web server for prediction of multiple level function of certain protein.Structure defines the function of a protein.Through structural homologies functional insights from a known protein are transferred to target proteins by the COFACTOR server.For the function prediction it combines global and local comparison algorithm. Its outputs include protein–ligand binding interaction, Enzyme Commission, and Gene Ontology (GO) [44]. COFACTOR also predicts structural analogs and functional homologs.
Mutational Analysis of Rv1165
Change in a single amino acid can result in disability of protein or loss of activity of protein which may hinder cellular processes.Mutational analysis can be exploited for design of experimental procedures in formulating anti tuberculosis drugs.Structure and sequence based mutation is important as it provides the comparison between the outcomes.Server used for mutation based structure and sequence is I-Mutant Suit 3.0.It is exploited for prediction of changes in stability of protein upon single point mutation.I-Mutant is based on support vector machine (SVM) [45]. I-Mutant Suit 3.0 groups the results into three classes: neutral mutations ranges from-0.5 ≤ ΔΔG ≥ 0.5 kcal/mo), deeply decreasing mutations ranges from <−0.5 kcal/mol, and mutations responsible for vast increasing in stability (>0.5 kcal/ mol).The free energy change (ΔΔG) anticipated by I-Mutant Suit 3.0 depends on the distinction amidst change in unfolding Gibbs free energy of mutant and native protein (kcal/mol) [46].
Results
Multiple Sequence Alignment
The sequence of target protein is retrieved using Mycobrowser which contains all the proteins of M. tuberculosis which are aligned with the same gene in different bacterium such as Escherichia coli K-12, Azospirillum brasilense, Moraxella catarrhalis BBH18, Shigella flexneri, Pseudomonas aeruginosa (P. aeruginosa) that have the same gene.The target protein Rv1165 of M. tuberculosishas 99 % homology with typA of P. aeruginosa (as shown in Figure 1).
Clustal omega is used for alignment of TypA gene of Mycobacterium tuberculosis H37Rvwith other organism such as Escherichia coliK-12, Azospirillum brasilense, Moraxella catarrhalis BBH18, Shigella flexneri, Pseudomonas aeruginosathat have the same gene.
Protein-Protein & Protein-Compound Interaction
String:Proteins present in the cell environment interact with each other. Interactions among the proteins present in cell environment are studied using STRING.STRING v 9.1 is a search tool for retrieval of interacting genes.STRING database shows results such as functional interactions among the proteins, stable complexes.The results are depicted on the basis of C-score.The cutoff ranges from 0-1.The low confidence: scores <0.4; medium: 0.4 to 0.7; high: > 0.7. Rv1165 interacts with Rv1163, Rv1164, Rv2316 and Rv3442c.According to the outcomes of STRING database server Rv1165 has highest interaction with Rv1163 (narJ) which is a respiratory nitrate reductase delta chain with confidence score i.e. 0.801 and other proteins have a confidence score ranging between 0.6- 0.7.Other interacting partners are also predicted as shown in Figure 2 & Table 1.
String tool predicts the interactive protein with proficiency of interaction. The figure shows that Rv1165 interacts narJ (Rv1163) with 0.801 score and narI (Rv1164) with 0.792 score and the cutoff of the server is 0-1.
The interacting partners are enlisted using STRING database which shows that interacting partners of Rv1165 protein of M. tuberculosis with other proteins like narJ, narI, polA, rpsl, uspA.
Stitch: For the prediction of ligands that interact with target protein STITCH database is used.Interaction studies done by the database includes both direct (physical) and indirect (functional) associations.STITCH database interactions are derived from 5 sources such as genomic context predictions, high-throughput lab experiments, co- expression, and automated text mining and previous knowledge in databases.The C-score ranges from 0-1 where the confidence score 1 denotes highest possible confident shows the compounds that interact with target protein are sulphate, phyrophosphate, GTP, MgADP, MgATP, 3’-phosphoadenosine 5’-phosphosulfate, phosphosulfate, distilled water, adenosine monophosphate and guanosine diphosphate.The highest confidence score is 0.996 which is of sulphate and other compounds have confidence score ranges from 0.859-0.893 and other ligands as shown in Figure 3.
Stitch tool predicts the interactive ligands which conclude that Rv1165 interacts with ligands such as sulphate, pyrophosphate, guanosine triphosphate, distilled water and other ligands. Sulphate has highest C-score that is 0.966 and pyrophosphate 0.893 and guanosine triphosphate 0.874 and the cut off is 0-1.
Ab Initio Modelling Via I-Tasser
The protein has 628 amino acids with UniProt ID O06563.Homology modeling for the target protein is done using servers such as RaptorX, Phyre2, I-TASSER, Swiss Model, LOMETS. Satisfactory results are obtained from I-TASSER.Threading programs used by I-TASSER include MUSTER, FFAS-3D, SPARKS-X, HHSEARCH2, HHSEARCH I, Neff-PPAS, HHSEARCH, pGen THREADER, wdPPAS and PROSPECT2.Results produced by threading template shows that Rv1165 protein, PDB hit is 4zciA and 3cb4D whose identity score 42% and coverage area of the template is 89% and normalized Z-score is 4.66 for 4zciA.The C-score which is the certainty score ranges from -5 to 2.For the model built by I-TASSER for Rv1165 the C-score is 1.15, the recorded TM score is 0.87±0.07 and the RMSD is 5.3±3.4Å which is good enough as shown in Figure 4.
Rv1165 protein in model is constructed by I-TASSER C-score is 1.15 ,estimated score is 0.87±0.07 and TM score is 5.3±3.4Å. (a) Sticks model, (b) cartoon model, (c) dots model, (d) ribbon model.
Validation of Predicited Model of Rv1165
Verify 3D: For the evaluation for 3D structure of the target protein verify 3D is employed. Nearest proximity of an atomic model is assessed with its own specific amino acid arrangement in 1D.Each deposit has crucial zone and condition (alpha, beta, circle, polar). Range of the score is between -1 to +1. 83.44% of the arrangement had found in the centre estimation of 3D-1D score >=0.2 that is that is satisfactory.
Errat: ERRAT favors the protein structure in the commencement of atomic alliance between different sorts of molecules.The quality factor is 89.0145% which is satisfactory.
Rampage: Model evaluation by SAVES Meta server is done by RAMPAGE (Ramachandran plot analysis).It is online server which shows that 74.5% residues are in most favored region, 21.2% in allowed region and 1.5% in outer region.This predicts that protein model is acceptable as shown in Figure 5.
RAMPAGE analysis shows that in favored region there were 74.5% of residues and in allowed region 21.2% of residues and outlier region 2.8% of residues were present.
Ligand Binding Pockets
Ligand binding pocket is predicted by COACH server.COACH server utilizes 2 strategies for prediction of binding site that are TM-SITE and S-SITE.Results for ligand binding site by the COACH server forecast that Rv1165 binds to guanosine 3’, 5’-bis (diphosphate), magnesium and nucleic acids.By assessment of the COACH server outcomes suggests that the rank 1 PDB hit is 4zcmA which has C-score 0.69 where the cluster size is 54, ligand name is G4P which is guanosine 3’, 5’-bis (diphosphate) and the consensus binding residues are 11, 12, 13, 14, 15, 16, 17, 18, 131, 132, 134, 178, 179 and 180as shown in Figure 6.
For the prediction of ligand binding site of Rv1165 has been done using Coach Server that predicts that Rv1165 binds to guanosine 3’, 5’-bis (diphosphate) at 11, 12, 13, 14, 15, 16, 17, 18, 131, 132, 134, 178, 179 and 180.
Functional Prediction
COFACTOR forecast the functions of the query gene Rv1165.COFACTOR predictionsincludes results such as structural analogs in PDB format, molecular function, biological process, cellular component and enzyme homolog in PDB format.Among the top 10 structural homologs the first ranked homolog has PDB hit 4zciA which is a crystal structure of E. coli GTPase BipA/TypA which has a TM-score of 0.89, RMSD is 0.37 and identity is 46.2% and coverage is 0.8888.Molecular function forecasted by the server suggests that the target gene TypA has 99% catalytic activity with C-score0.99 and 98% GTPase activity.According to biological process typA is involved in cellular process. C-score for cellular process is 1.00 as shown in Figure 7A, B & Table 2.
The figure shows gene ontology term with molecular function and biological process. The whole procedure demonstrates the C-score of 0.99 with the catalytic activity and 0.98 with GTPase activity and mainly involved in cellular process with C-score 1.00.
Functional analysis of target gene is done using COFACTOR which shows that its 99% molecular function is catalytic activity, 100% works in cellular process and is a cellular component.
Mutational Analysis of Rv1165
Proteins are composed of amino acids and altering a single amino acid can result in change in the stability of protein structure. I –Mutant 3.0 is used for analyzing the stability of the protein upon changes at single nucleotide positions.The positions for mutation are selected upon the fact that amino acid those are different in TypA gene of M. tuberculosisfrom TypA gene of Escherichia coliK-12, Azospirillum brasilense, Moraxella catarrhalisBBH18, Shigella flexneri, Pseudomonas aeruginosa that have the same gene.DDG value predicts the stability.The DDG value less than -0.5 predicts large stability decrease and value greater than 0.5 increases the stability of the protein and value between -0.5 to 0.5 predicts neutral stability.In target protein Rv1165 stability is largely decreased at the position 329 where the wild type amino acid is valine which is changed to methionine and the DDG value is -1.13 and at the position 371 where isoleucine is changed to threonine and DDG value is -2.14.Trivial change is stability is predicted at different positions as shown in Table 3.
Study of protein stability change after a single point mutation by using I- Mutant 3.0.The table concludes that the stability highest decreases stability at 371st residue by changing isoleucine into threonine.
Discussion
With the emerging world jeopardy rooted from Tuberculosis are also mounting.Although there are various treatment and drugs regimens against the malady it is most menace aliment among all.Drugs and vaccine such as Bacilli-Calmette-Guerin are poignant towards ineffectiveness because M. tuberculosis strains are being transformed into Multi-Drug Resistance, Extensively-Drug Resistance strains.Thus there is a pressing need to uncover conducts that can bring a standstill to the transmission of the alignment.In this manuscript we have exploited insilico approaches against typA (Rv 1165) gene of M. tuberculosis. Main function of the gene is that it is a possible GTP binding translation elongation factor typA (tyrosine phosphorylated protein A). Rv1165 (typA) contains concord sequence such as DXXG, GXXXXGK motifs as a probable GTP-binding protein.Enormous insilico approaches have been applied to detail various function and essentiality in biological process.Rv1165 of M. tuberculosis is found to be 94% similar to Pseudomonas aeruginosa, and other organisms such as have sequence similarity.Interaction studies done by STRING database shows that as typA is GTP binding protein, its interacting functional partners are important.Itinteracts with other genes that areRv1164, Rv1163 that are probable narI and narJ respectively.Both are respiratory nitrate reductase [30-33]. Study shown by STITCH which shows interacting ligand predicts that Rv1165 interacts with sulphate, phyrophosphate, GTP [34-35]. Valid model for the target protein is built by I-TASSER (as shown in Figure 4) [36-37] and validated by SAVES Meta server (ProSA, ProQ, Verify3D, Rampage) where model evaluated by Verify 3D is satisfactory [38-39]. Binding sites predicted by COACH proves that Rv1165 binds with guanosine 3’, 5’-bis (diphosphate), magnesium and nucleic acids as shown in Figure 6 [40-42]. Functional analysis performed by COFACTOR states that its molecular function in catalytic process and biological function is in cellular process (as shown in Figure 7A, B [43-44].Mutations are done on points where amino acids are different from typA of different organism. Result from I-Mutant shows that highest decrease in stability occurs at 329 and 371 position.At 329th position wild type amino acid valine is replaced by methionine and at 371 positions where isoleucine is replaced by threonine [45-46]. As discussed above insilico approaches are important step in knowing the cellular function of the bacterium and therefore could be helpful in understanding more about the bacterium and finding cures against the malady.
Conclusion
We need to stop the escalating effect of Tuberculosis worldwide.Therefore, it is important to know about each aspect.This study concludes with the knowledge of some important aspects of typA (Rv1165) gene and interacting partners to know about certain pathways.For the betterment of the knowledge about the gene this manuscript provides initial understanding which might help in designing in vivo and in vitro experiments.
Acknowledgment
The author acknowledges financial support from the Department of Science and Technology-SERB, Council of Scientific and Industrial Research-Institute of Genomics and Integrative Biology under the research project GAP0145.

Figure 1: Sequence Alignment by Clustal Omega.
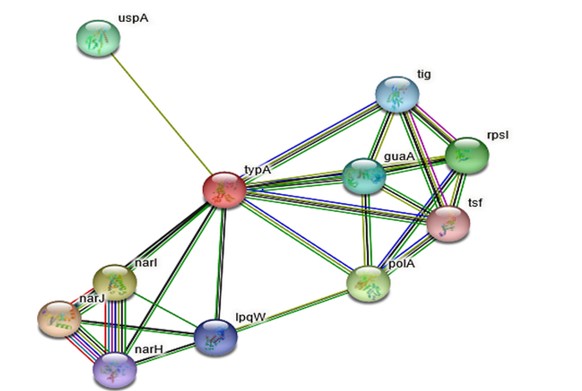
Figure 2: Protein-Protein Interaction of Rv1165 by STRING Tool.
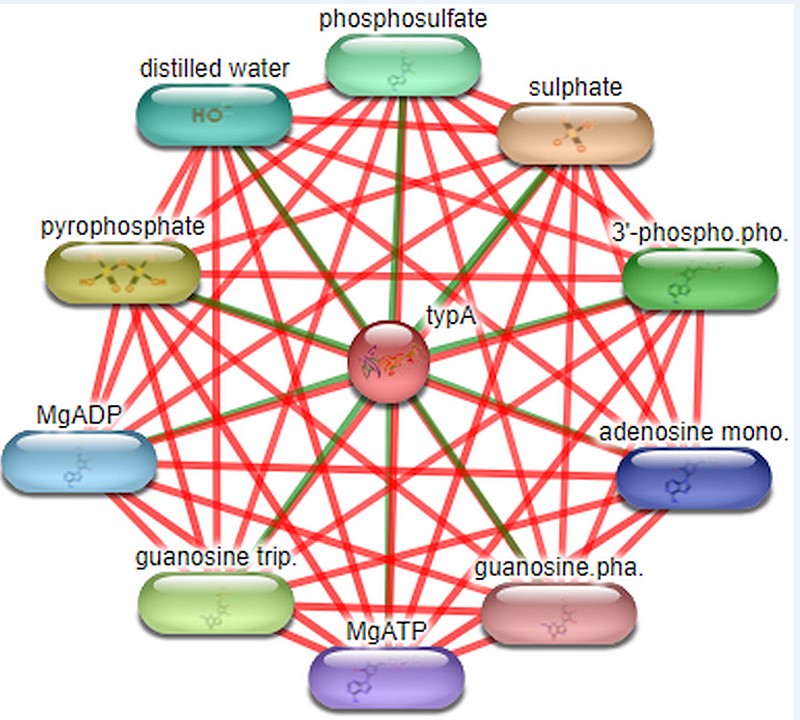
Figure 3:Protein-compound interaction of Rv1165 by STITCH tool.

Figure 4:Modelling of protein by I-TASSER.
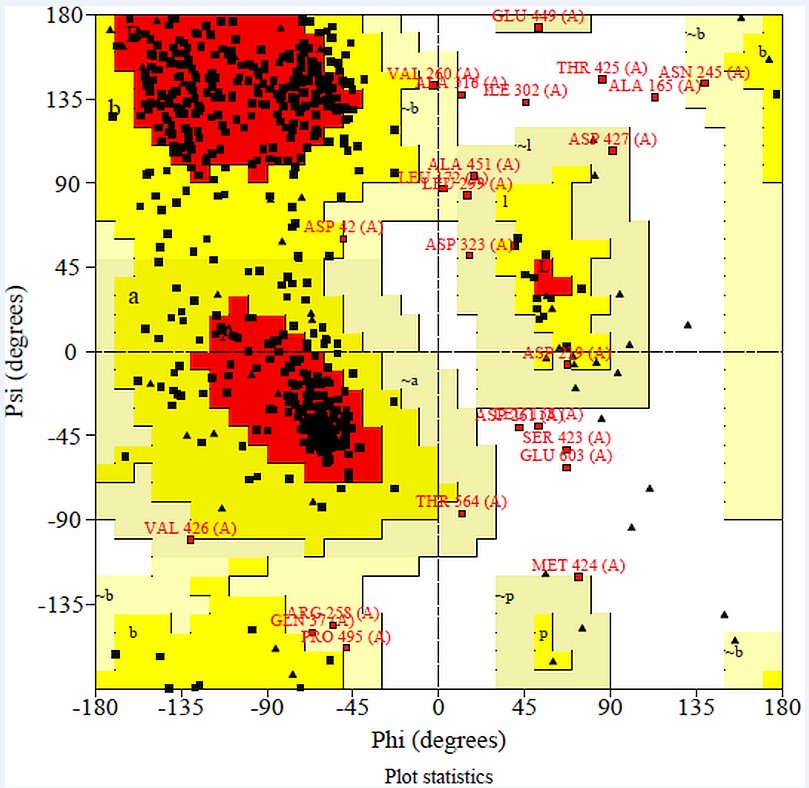
Figure 5: RAMPAGE analysis of modeled protein.
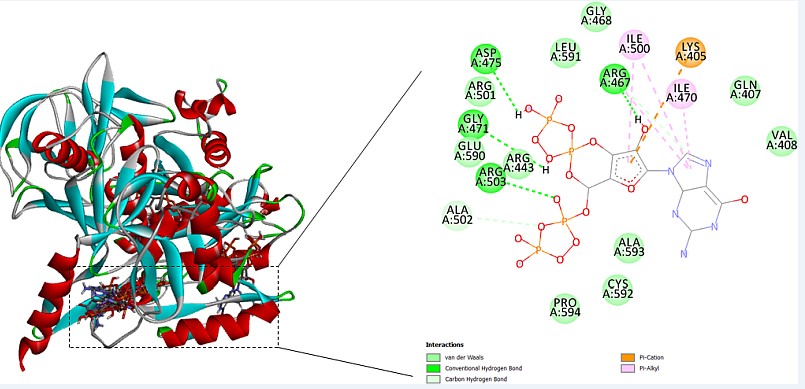
Figure 6:Prediction of ligand binding site.
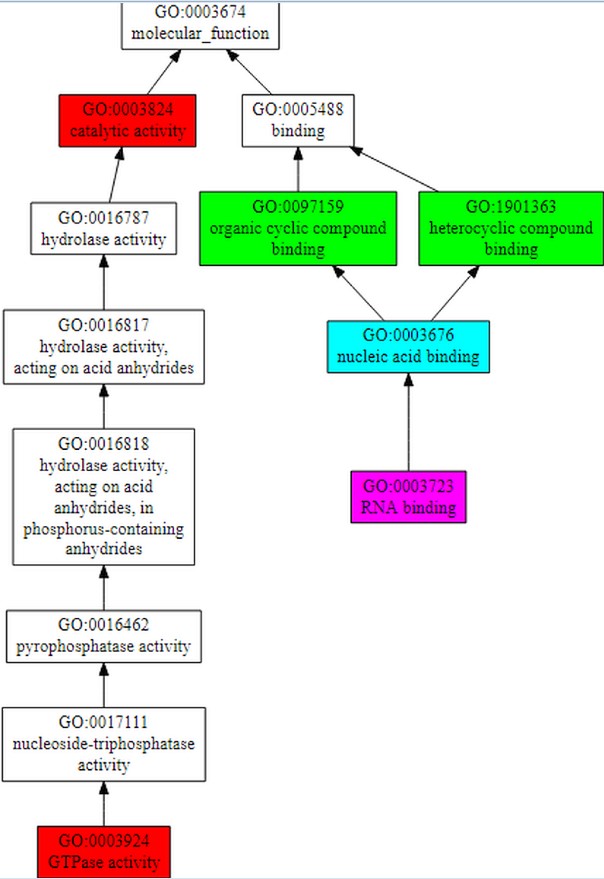
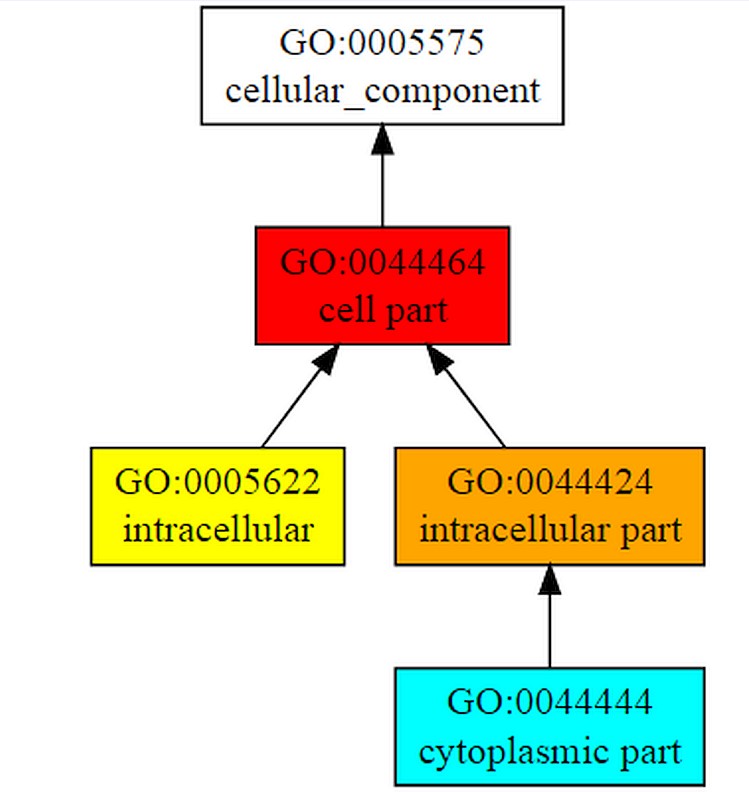
Figure 7A, B: Structure-based function prediction by cofactor server.
|
Sr. No. |
Functional Partners |
Function |
Score |
|
1. |
narJ |
Respiratory nitrate reductase delta chain |
0.801 |
|
2. |
narI |
Respiratory nitrate reductase gamma chain |
0.792 |
|
3. |
polA |
Polymerase activity |
0.741 |
|
4. |
Rpsl |
Ribosomal protein |
0.739 |
|
5. |
uspA |
Sugar-transport , ABC transporter |
0.660 |
|
6. |
guaA |
Catalyzes the synthesis of GMP from XMP |
0.654 |
|
7. |
Tig |
Protein export |
0.638 |
|
8. |
IpqW |
Regulates the lipoarabinomannas (LAMs) synthesis |
0.635 |
|
9. |
narH |
Respiratory nitrate reductase beta chain |
0.634 |
|
10. |
Tsf |
Induces exchange of GDP to GTP |
0.614 |
Table 1: Table for interacting functional partners.
|
Sr. No |
Molecular function |
Biological process |
Cellular component |
|||
|
GO term |
C-score |
GO term |
C-score |
GO term |
C-score |
|
|
1. |
Catalytic activity |
0.99 |
Cellular process |
1.00 |
Cellular part |
1.00 |
|
2. |
GTPase activity |
0.98 |
Cellular metabolic process |
0.98 |
Intracellular part |
0.84 |
|
3. |
Heterocyclic compound binding |
0.70 |
Organic substance metabolic process |
0.97 |
Intracellular |
0.72 |
|
4. |
Organic cyclic compound binding |
0.70 |
Organic substance biosynthesis process |
0.93 |
Cytoplasmic part |
0.52 |
|
5. |
Nucleic acid binding |
0.53 |
Cellular biosynthesis process |
0.93 |
- |
- |
|
6. |
RNA binding |
0.50 |
Cellular macromolecule metabolic process |
0.92 |
- |
- |
Table 2: Table for functional analysis.
|
I-Mutant 3.0 |
||||
|
Position |
Amino Acid In Wild Type Protein |
New Amino Acid After Mutation |
Ddg Value |
Prediction Effect |
|
30 |
L |
F |
-0.92 |
Decrease |
|
69 |
T |
N |
-0.79 |
Decrease |
|
171 |
G |
D |
-0.88 |
Decrease |
|
209 |
K |
D |
0.46 |
Decrease |
|
216 |
G |
V |
-0.35 |
Decrease |
|
249 |
R |
K |
-0.83 |
Decrease |
|
329 |
V |
M |
-1.13 |
Decrease |
|
371 |
I |
T |
-2.14 |
Decrease |
|
483 |
S |
T |
-0.32 |
Decrease |
Table 3: Table for mutational analysis.
Citation: Meena S, Bhanawat S and Meena LS (2019) Insilico Analysis and Homology Modeling of TypA (Rv1165) Protein of Mycobacterium Tuberculosis H37Rv. Curr Trends Biotechnol Biochem: CTBB-100008.
