
Current Trends in Biotechnology and Biochemistry
Research Article
Prescience of Rv1786 of Mycobacterium Tuberculosis H37 Rv to Assist the Pathogenic Potentials and Significance by Computational Annotations
Meena S, Agarwal P, Mishra M, and Meena LS*
CSIR-Institute of Genomics and Integrative Biology, India
*Corresponding author: Laxman S. Meena, CSIR-Institute of Genomics and Integrative Biology, India, Tel: 011-27666156; Email: meena@igib.res.in
Citation: Meena S, Agarwal P, Mishra M, and Meena LS (2019) Prescience of Rv1786 of Mycobacterium tuberculosis H37Rv to assist the pathogenic potentials and significance by computational annotations. Curr Trends Biotechnol Biochem: CTBB-100006
Received date: 21 August, 2019; Accepted date: 27 August, 2019; Published date: 04 September, 2019
Abstract
Globally, tuberculosis (TB) is one of the major reasons of increased mortality rate according to WHO. We are presenting probable information for treatment and research against this disease. Rv1786 gene is a probable ferredoxin protein which is involved in intermediary metabolism or respiration of Mycobacterium tuberculosis H37Rv (M. tuberculosis). Ferredoxins are iron-sulphur proteins that mediates electron-transfer in cytochrome P450 mono-oxygenase. Electron-transfer is elemental for the existing entities, which participate in respiration to generate chemical energy inside cell milieu. Using some bioinformatics approaches, this protein is found to be a cytoplasmic protein having role in iron binding, electron transfer and showing catalytic activity. Molecular docking reveals interaction of this protein with the compounds 3 g2n, AdoHcy, D-Gap and S-Adenosyl M which can be used to target this gene against tuberculosis. This manuscript noted some of the most important aspects about hypothetical protein Rv1786 being functional as ferredoxin protein. The major insights of this study include multiple sequence alignment, interaction study, Ab-initio modelling by QUARK server, validation of the model by RAMPAGE analysis, prediction of metal binding, structure is validated using Insilico approaches. Interaction study by STITCH server shows that this protein interacts with its neighbouring partners. Prediction of ligand binding site confirms that this gene binds with sulphur tetra fluoride by which we can predict this gene as ferredoxins protein which is essential for pathogenesis. This study will be helpful in developing new drug targets against this gene for the treatment of this disease.
Keywords: Cytochrome P450; Mycobacterium tuberculosis H37Rv; Ferredoxins
Abbreviations
AIDS : Acquired Immunodeficiency Syndrome
HIV : Human Immunodeficiency Virus
M. tuberculosis : Mycobacterium tuberculosis H37Rv
M. smegmatis : Mycobacterium magmatism
NCBI : National Centre for Biotechnology
MDR-TB : Multidrug-Resistant TB
MSA : Multiple Sequence Alignment
SDG : Sustainable Development Goals
TB : Tuberculosis
RAMPAGE : Ramachandran Plot Analysis.
WHO : World Health Organisation
XDR-TB : Extremely Drug-Resistant TB
Introduction
Mycobacterium tuberculosis H37Rv (M. tuberculosis) strain is highly pathogenic strain among the Mycobacterium species. It is a gram-positive, non-motile, obligate aerobe, having doubling time of approximately 24 hours and non-spore developing bacilli. Due to its complex nature of cell wall, macrophage cannot completely eradicate the bacterium and also this bacillus inhibits merging of phagosomes with lysosomes [1-3]. Pathogenic strain H37Rv of this bacterium is different from other non-virulent strains like Mycobacterium smegmatis (M. smegmatis) [4]. It is a single infectious agent which asserts many lives (co-infection with HIV/AIDS). World Health Organisation (WHO) declares that the tuberculosis is a major cause of mortality now a day’s [5]. Millions of people, continuously developing tuberculosis (TB) each year and 2017 records have shown that 1.3 million people among HIV-negative people died due to TB and there were an additional 300 000 deaths from TB among HIV-positive people. Globally, the best estimate is that 10 million people developed TB disease in 2017 among which 5.8 million were male; 3.2 million were female and 1 million were progenies [6]. TB is a serious bacterial communicable infection that mainly affects respiratory part and the condition is known as pulmonary tuberculosis but if it is not treated completely on time, it can also affect the other body parts like brain, urinary tracts, bone etc. known as extra pulmonary tuberculosis (EPTB) [7]. Pulmonary tuberculosis is common while EPTB is also a current clinical problem. In spite of having Bacille Calmette-Guerin (BCG) vaccine and the (DOTS) effective short-course chemotherapy, the exposure of drug-resistant strains with the deadly combination of the HIV is only reason of increased patients of TB among developing countries [8]. Initial medicine for TB are first line drugs (FLD) such as Streptomycin, Ethambutol, Pyrazinamide, Rifampicin and Isoniazid and these are very effective for the treatment of TB but these drugs are now unproductive to treat TB due to drug resistance in bacteria. The emergence of Multidrug-Resistant-TB, i.e. resistance for Isoniazid and Rifampicin, requires the use of second-line drugs which are not easy to acquire and are costly or toxic than FLDs [9]. That’s why for the inhibition of MDR-TB it is essential to invent and implement of drug resistant TB [10]. Genomic research & exploration find an assortment of effective medicines which may aid in the treatment of diverged TB such MDR, XDR and TDR. These new types of tuberculosis prompt us to move to form modern drugs. M. tuberculosis is a self-seeking pathogen of alveolar macrophages. A person having weak immune system can get easily infected by this bacterium [11]. Insilico approaches provide the information and knowledge which will be helpful in generation of novel therapies to deal with this air born infectious diseases. In this manuscript, we are dealing with the detailed characteristics of Rv1786 with the help of Insilico techniques [6]. It is estimated that it may act as ferredoxins like protein. Rv1786 is assumed ferredoxins. Ferredoxins (iron-sulphur proteins) mediates electron transfer in cytochrome P450 mono-oxygenase (CYP)-related catalytic reactions in a wide-ranging diversity of organisms. Electron transfer is an elemental process for all existing entities, which involved in respiratory processes to generate chemical power inside the cell [12]. Rv1786 is determined by a gene located downstream of the gene encoding CYP143A1 in the genome of M. tuberculosis [13]. Contrarily, the structure and function of Rv1786 is remained undetermined. M. tuberculosis encodes for twenty cytochrome P450 enzymes (CYPs), well thought-out possible drug-hits due to their decisive roles in bacterial achievability and host bug. Catalytic action of mycobacterial CYPs is dependent on electron transfer from NAD (P) H-ferredoxins-reductase (FNR) and ferredoxins (Fds). Mycobacterium genome contains 2 FNRs (FdrA and FprA) and 5 ferredoxins (Fdx, FdxA, FdxC, FdxD, & Rv1786). Though, the related redox coalition has not been fully recognized [14]. As the information about the protein Rv1786 is not completely revealed so by using some bioinformatic tools we have studied our query about the probable ferredoxin protein Rv1786.
Material and Methods
There are many servers and bioinformatics tools used for Insilico study and few of them we have used here to study our protein Rv1786. Living sequences like proteins or nucleic acids which are of same stretches are aligned in the group of three or more to study or surmise the homology and the evolutionary relationships among sequences is known as Multiple Sequence Alignment (MSA) for which, Pairwise Sequence Alignment tools are used to recognise regions of resemblance that may stipulate structural, functional or evolutionary relationships between two biological sequence [15] is a multiple sequence alignment program (Consistency-based MSA tool) that attempts to mitigate the pitfalls of progressive alignment methods. Its main characteristic is that it will allow you to associate results obtained with many alignment methods. This tool can align up to 500 sequences or a maximum file size of 1 MB [16]. Multiple sequence alignment (MSA) approach for ferredoxin protein analysis (ferredoxin gene Rv1786) of M. tuberculosis is done with ferredoxins gene of Mycobacterium bovis (M. bovis), Mycobacterium tuberculosis erdman Mycobacterium marinum (M. marinum), and M. smegmatis [17].
Multiple Sequence Alignment
Prediction of the interacting functional partners: Interaction Study is done by STITCH for the demonstration of protein-protein and chemical-protein association between two or more existences. A protein may work in the web like manner and collaborate with different proteins in the cell cytoplasm. The associations incorporate direct (physical) and indirect (functional) interactions. The application of STITCH is the computational prediction which is based on the sequence interrelation between the entity, and from connections aggregated from other databases [18]. STITCH database interaction result shown unconnectedly for data and the cut-off value as low confidence: scores <0.4, medium 0.4-0.7 & high >0.7 [19].
Protein subcellular localization prediction: PSORTb version 3.0, most accurate bacterial SCL estimate software got recognition in 2005, and has been commonly used for the SCL prediction of individual proteins as well as for whole proteomes. It generates prediction results for five major localizations for Gram-negative bacteria (cytoplasmic, inner membrane, periplasmic, outer membrane and extracellular) and four localizations for Gram-positive bacteria (cytoplasmic, cytoplasmic membrane, cell wall and extracellular) [20].
Disulphide Bond Predictions: Disulphide bonding helps in the determination of experimental structure and interprets the solidity of the proteins. Disulfide bonds in protein shows vital role in protein stability conformation. Disulfide bridges are formed by cysteine residue in the primary structure of proteins [21]. Disulfide bonds and Cysteine residues are essential for protein composition and function. Due to the utilization of SVM binary server to DiANNA 1.1 web server predict bonding state of cysteine, this cysteine is paired by Recursive Neural Network to show di-sulphide bridges [22].
Epitope Prediction: Primary sequence of (Rv1786) ferredoxin protein was used for the prediction of B-cell & T-cell epitopes for which we have used online bioinformatics tools. ABCpred and BCpred tools were used for B-cells and T-cell estimation and PROPRED server was used to predict MHC II binding epitope [23].
Ab-Initio Protein Folding: QUARK is used to predict protein structure and function. QUARK is a computer based procedure for ab-initio protein folding and protein structure prediction, which aims to construct the accurate protein 3D model from amino acid sequence only. QUARK models are constructed from small fragments (1-20 residues long) by replica-exchange Monte Carlo simulation under the guide of an atomic-level knowledge-based force field. QUARK was ranked as the No 1 server in Free-modelling (FM) in CASP9 and CASP10 experiments. Since no global template data is used in QUARK simulation, the server is appropriate for proteins which are considered without homologous templates [24].
Structure Prediction: The targeted protein Rv1786 (ferredoxin) is analysed and endorsed by the SAVES meta server (version 4) which have RAMPAGE, Verify3D and ERRAT for the rectification of protein. SAVES-meta server checks the stereochemical quality of a protein structure by analyzing residue-by-residue geometry and whole structure geometry demonstrated the protein validate score for the validated result in most favoured region, additionally allowed region, generously allowed region & disallowed region [25, 26]. This metaserver tracks six programs for checking and validating protein structures during and after model rectification. In our model refinement we use RAMPAGE, Ramachandran Plot Analysis is based on an assessment of ramachandran plot. The RAMPAGE server favours the protein structure on the premise of ????, ???? point of individual deposits [27,28]. Verify3D the 3D structure of the protein was contrasted with its own amino acid sequence thinking about a 3D profile ascertained from the nuclear directions of the structures of correct proteins [29,30] and the general characteristics of the demonstrated structures were evaluated by the ERRAT [31].
Ligand Binding Prediction: Prior to vibrant site exact binding analysis (molecular docking); the pledge of authentic pocket is important. Binding pocket is the spot of the protein where the small ligand can reversibly or irreversibly bind. Just a pair of amino acid deposits is in charge of the ligand. However other amino acids build-ups of protein are given accurate preface and assertion for the ligand binding site prospect loom for COACH server. Ligand binding prediction of our displayed protein has been done by COACH online server. COACH-mega server forecast protein-ligand binding sites from given starting structure of said proteins by using TM-SITE and S-SITE methods COACH generates corresponding ligand binding site predictions, which recognise ligand-binding templates from the BioLiP protein function databases by binding precise substructure and sequence profile examinations. In COACH server, output has ranked top10 model by the clump size, given C-score, PDB hit, ligand name, complex structure download, and consent lees range values of C-score prediction lie between 0 and 1, where the uppermost score display more reliability. The majority of the proteins (=90%) can be modelled with a correct fold (TM-score>0.5) and 65% have a RMSD below 6°A, although all close homologous templates were expelled in the model generations [32].
Structural-Based Functional Analysis: Automated structure based protein function annotation is done by COFACTOR webserver. Starting from a structural model, given by both computational modelling and investigational determination, COFACTOR first identifies template proteins of analogous folds and effective sites by threading the target structure through three representative template libraries that have known protein–ligand binding connections, Enzyme Commission number(EC) or Gene Ontology(GO) terms [33]. COFACTOR was ranked as the best method for protein–ligand binding site estimations. Inspection of this protein functioning for threading was done by BioLiP protein work database. By this database, analysis of the model structure tells us how to observe the homology and the efficient state of the protein. COFACTOR model-based capacity expectation evaluation was cited as the best method for protein, determines protein structure composition [34].
Molecular Docking: Molecular docking of Rv1786 was done by using HDOCK webserver, a novel web server that supports protein-protein and protein-DNA/RNA docking and agrees both sequence and structure of proteins. Protein-DNA/RNA and protein-protein interaction play an elementary role in a range of biological processes. Formative the complex structures of these connections are worthless in which molecular docking has played an important role [36].
Results
Multiple sequence alignment: T-COFEE server was used for multiple sequence alignment of our targeted protein Rv1786 of Mycobacterium tuberculosis H37Rv with other species of the bacteria like Mycobacterium bovis, Mycobacterium tuberculosis erdman, Mycobacterium marinum, and Mycobacterium smegmatis. First we retrieve sequence in FASTA format of protein the server aligns sequence and gives the perfect matches. The percent identity matrix proves its identity among all species. The similarity between all species is might be an indicator for its importance of the process in the survival of these prokaryotes. The MSA result shows score 972 for our input sequences and the pink colour corresponds to alignment portions with a strong support in the primary library as shown in Figure 1.
Prediction of the interacting functional partners: STITCH database server result depicts that Rv1786 (Ferredoxin) is a cytoplasmic protein interacts with S-adenosylmeth, AdoHcy, cobalt, Co(NH3)6, Rv3198.1, cyp143 and ppe25 which shows that the interaction result with other proteins has minimum interaction score [between 0.4 & 0.6] shown in the Figure 2 and Table 1, but it shows high interaction score for S-adenosylmeth, i.e., 0.795.
Protein subcellular localization prediction: According to PSORTb server, Localization Scores for Cytoplasmic localization is 7.50, for Cytoplasmic Membrane is 1.15, for Extracellular is 0.73 and for Cell wall is 0.62. Final Prediction for the protein localization is Cytoplasmic as the score for cytoplasmic localization is highest (7.50). So, Rv1786 is a cytoplasmic protein, exist in the cytoplasmic region with final predicted localization score 7.50.
Disulphide Bond Prediction: For the Di-sulphide bond analysis of ferredoxin protein (Rv1786), DiANNA 1.1 web server was used. As shown in Figure 3, Di-sulphide bonds are predicted on the position of 10th-16th, 10th-54th and 16th-54th with score value 0.01051, 0.01038 and 0.38842 respectively.
Epitope Prediction
B-cell epitope Prediction: ABC pred depicts numerous B-cell epitopes taking the overlapping window of 52 amino acids that results in the best probability score of 0.81 from the residues region “PEDMQLTRDGVAACPE” that starts at 41st residue as shown in Figure 4.
T-Cell epitope estimation: Multiple DR-β1 alleles were used like HLA-DRB1*0101, HLA-DRB1*0102, HLA-DRB1*0301 for the prediction of the T-cell epitopes with the MHC class-II binding region in the antigenic protein sequence of Rv1786. Two consensus epitopes were - IMTERCLSI and VGHAQCYAV in (DRB1_0101, HLA-DRB1*0102) at the 09th - 17th and 36th-44th residue positions, and four consensus epitopes – LSISHRVRV, VRLDPSRCV, FPIDDSGNS and LTRDGVAAC were observed in DRB1*0301 at 15th-23rd, 28th-36th, 49th-57th and 70th-79th residue positions respectively in sequence as their respective alleles at 3% threshold as revealed in T-cell epitope prediction, as shown in Figure 5.
Ab-Initio Protein Modelling: Rv1786 protein was modelled by QUARK to predict structure and function. According to this, 82.5% residues are in most favoured region, 15.8% residues are in additional allowed region 1.8% residues are in generously allowed region and 0.0% residues are in disallowed region so it could be said as good model. ERRAT value is 87.7458 for our protein which is acceptable. Verify 3D score value is 86.57 which show PASS for this model as shown in Table 2.
Validation of Protein Structure
After modelling, the protein structure was validated by using SAVES server (RAMPAGE (Ramachandran Plot Analysis), ERRAT, and Verify3D). RAMPAGE Analysis: The targeted protein was validated by RAMPAGE server (Ramachandran Plot Investigation). After examination of Ramachandran Plot of our protein, structure demonstrated that 82.5% of residues have been present in a favoured region, although other 15.8% of residues were present in the additional allowed region and 1.8% was present in generously allowed region. ERRAT: - ERRAT server favours the protein structure on the premise of the nuclear connection between various sorts of atoms. The overall quality factor of our protein structure is 87.7458 which is acceptable. Verify3D: - The Verify3D strategy evaluates protein structure by utilizing 3-D profiles. This program examines the similarity of a nuclear model (3D) with its own certain amino acid sequence which is 1-dimensional. Every deposit doled out a basic class in radiance of its area and condition (alpha, beta, circle, polar, nonpolar, and so on). The score ranges from -1 (poor score) to +1 (good score). 86.57% of the sequence was found in the middle value of 3D-1D score >=0.2 that is perceptive for our demonstrated protein as shown in Table 2.
Ligand Binding Prediction: Rv1786 contains ligand binding site for Sulphur tetra fluoride that is confirmed by coach server. COACH results shows that binding of SF4 (Sulphur tetra fluoride) with this gene at rank 1 position has C-score of 0.99. SF4 binds with Rv1786 binding residue at 5,12,11,12,13,14,15,16,32,54,55,56,58,59 positions. COFACTOR result shows C-score 0.68, TM-SITE score 0.56, & S-SITE score 0.48.
Structure Based Function Prediction of protein: COFACTOR online server predicts function of protein on the basis of structure. It is a structure, organization, and protein-protein association based strategy for natural observation of protein particles. COFACTOR results predicted structural analogue in PDB, molecular capability, biological process, cellular segment, enzyme homolog in PDB, and outline protein with comparative binding sites. C-score GO is confidence score of predicted GO terms. GO terms for Rv1786 shows molecular function, biological process, cellular components result as shown in Figure 6. For molecular function, C-score of 0.84 is for electron carrier activity, 0.81 for iron, sulphur cluster binding,0.77 for catalytic activity and 0.77 for iron ion binding. For biological process C score is high for metabolic processes, single organism processes, and cellular processes. For cellular components, high C score values reveal that protein of Rv1786 lies in cytoplasm.
Molecular Docking: Docking for the gene Rv1786 is done by using HDOCK server with the compounds 3g2n, AdoHcy, D-GAP and S AdenosylM. 3g2n is binding to our protein by H bond, C-H bond and Pi-Alkyl bond with amino acid GLU36, ALA35 and ILE33. Docking score for this interaction is -4.6. AdoHcy is showing docking score -5.5 and the interacting bonds are H bond and Pi-Alkyl bond with interacting amino acids LEU5, ALA35, GLU36, HIS37, VAL39 and LEU34. D-GAP is showing score value -6.3 and interacting with ASP49, ARG48, VAL51, ALA52 and ILE60 with bonds H bond and Pi-Alkyl. S AdenosylM is showing score -4.8, interacting with LEU61, GLU63 and GLU62 with H bond, C-H bond and Pi-Alkyl as shown in Figure 7 and Table 3. These compounds can be used as targeting molecules against tuberculosis.
AdoHcy and 2D diagram analyse the interacting residues. (c) Rv1786 interacting with DGAP and 2D diagram analyse the interacting residues. (d) Rv1786 interacting with S-adenosylmethionine and 2D diagram analyse the interacting residues.
Discussion
Many years of past research, we have observed that tuberculosis asserts many lives. The risk of infection in healthy individuals is increasing day by day due to inapproachable drugs and treatments. There is no definite protective and therapeutic cure to overcome tuberculosis totally apart from BCG. In this manuscript, we accentuate Rv1786 of M. tuberculosis H37Rv. After computational analysis of this gene, we find that this gene is 7.35318 kDa molecular weight is probable ferredoxin proteins by Myco browser database [37]. MSA of this Rv1786 by T-COFFEE server confirmed that this protein shows similarity with other surrounding proteins which might be helpful for the further studies [38]. STITCH database shows that ferredoxin interacts with its adjacent genes like S-adenosylmeth with score 0.795 [39]. According to PSORTb server, Rv1786 is a cytoplasmic protein, present in the cytoplasmic region with final predicted localization score 7.50. Model forecast of this gene is designed by QUARK SERVER and validation of the model was completed by SAVES metaserver which confirms that the model of the protein has acceptable output [13]. Ligand binding was done by COACH SERVER and structure-based function prediction by COFACTOR server. Mutational analysis shows that this gene loses its stability in stress environments; therefore, further experimental studies on this gene might be beneficial to prove this gene as a new drug target.
Conclusion
We need an effective step to stop TB pandemic all over the world, so let’s zoom hands. As result of computational analysis of Rv1786, we find that this gene acts as Ferredoxin like protein which seems to be indispensable for the metabolic reactions in Mycobacterium tuberculosis H37Rv. This study concludes some important aspects and possible targets against the gene but further study might come out with the production of effective drug therapy against this tedious disease. Although the experiments enlisted in this manuscript are not enough for giving prediction for abolition of this hazardous disease, but these experiments may give a way towards better knowledge and initial steps in further in-vitro and in-vivo experimental works.
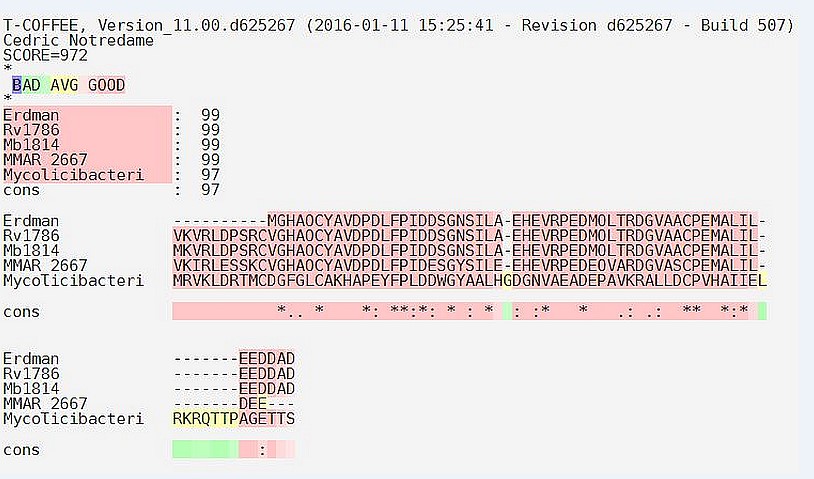
Figure 1: Systematic demonstration of the multiple sequence alignment by using T-COFFEE: The multiple sequence alignment of the Erdman strain, M. tuberculosis H37Rv, M. bovis, M. marinum and M. smegmatis with Rv1786 protein sequence result out the homology of this protein sequence with other proteins by using T-Coffee.
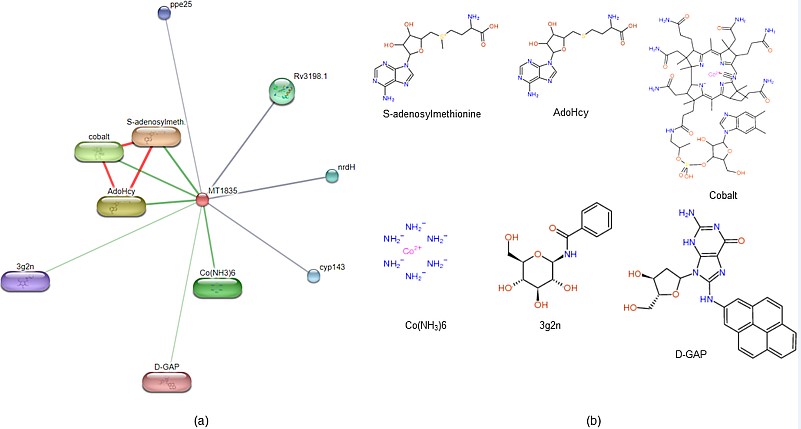
Figure 2: Protein-compound interaction: STITCH server predicts the interacting partner of the protein (a) protein compound interacting with Rv1786 are S-adenosylmethionine, AdoHcy, cobalt, Co(NH3)6, Rv3198, cyp143 and ppe25 (b) Rv1786 protein interacting compound structure are shown.
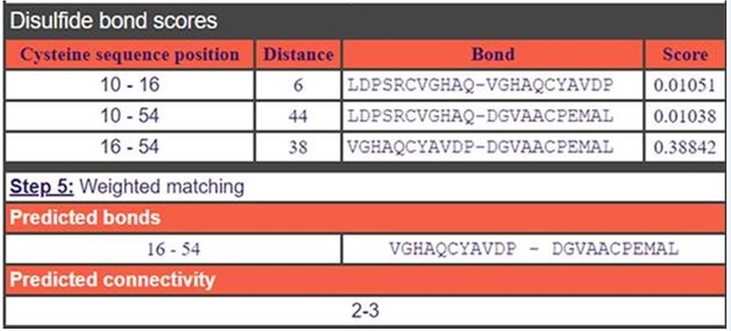
Figure 3: Disulphide bond prediction: In this figure, predicted di-sulphide bond are in between Cysteine-Cysteine residues. The figure clearly shows the Disulfide bonds at position 10-16, 10-54 and 16-54 with score values by using a trained neural network.
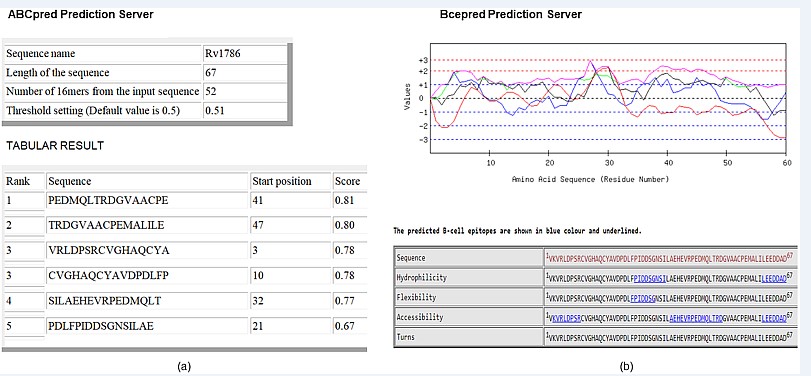
Figure 4: Showing the B-cell epitopes: In Rv1786 protein sequence predicted residue of (a) ABCpred predicted 5 epitopes at position 41, 47, 3, 10, 32 and 21 (b) BCEpred epitopes predicted by hydrophilicity (black colour), flexibility (blue colour), accessibility (green colour) and turn (red colour).
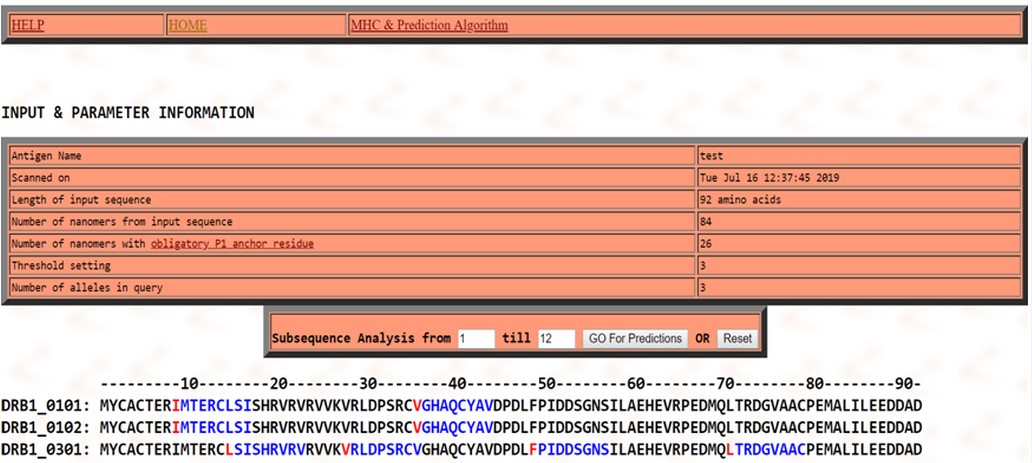
Figure 5: T- Cell epitope prediction: Two consensus sequence (shown in red & blue) were found for T –cell epitopes bind to MHC class II i.e. IMTERCLSI and VGHAQCYAV in DRB1_0101allele at 09th - 17th and in HLA-DRB1*0102 alleles at the and 36th-44th residue positions. Four consensus epitopes-LSISHRVRV, VRLDPSRCV, FPIDDSGNS and LTRDGVAAC were observed in DRB1*0301 allele at 15th -23rd , 28th -36th, 49th -57th and 70th -79th residue positions in sequence.
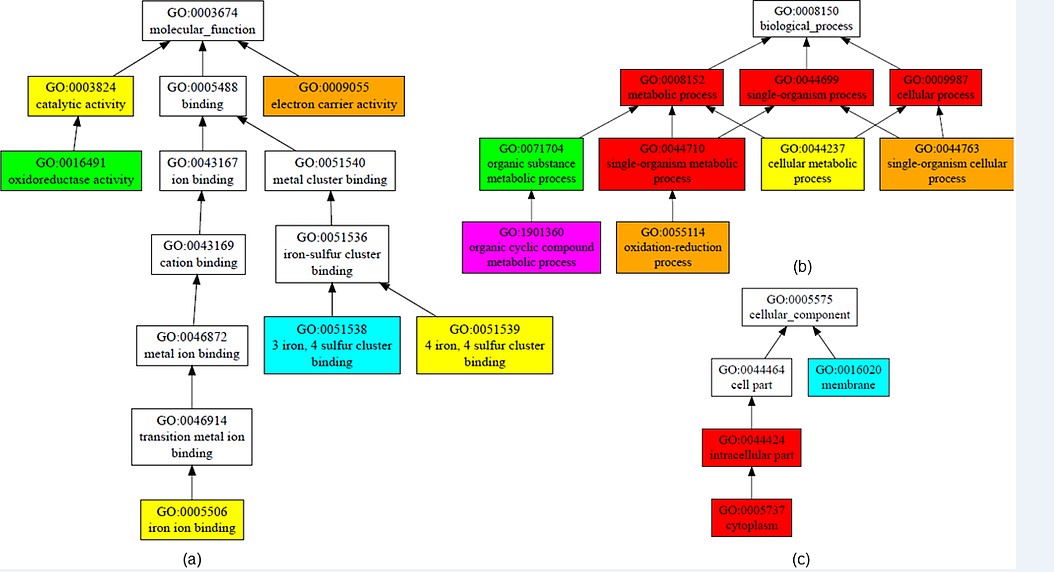
Figure 6: Structure-based function prediction: The figure shows gene ontology term with molecular function, biological process and cellular component. (a) GO term predicted molecular function with C-score 0.84 is electron carrier activity, 0.77 is catalytic activity and 0.77 iron ion binding. (b) GO term predicted biological process with high C-score for metabolic processes, single organism processes, cellular processes (c) GO term predicted cellular component is in cytoplasm. (pink colour denotes C- score of 0.4-0.5, blue green denotes 0.5-0.6, yellow denotes 0.7-0.8, orange denotes 0.8-0.9, Red denotes 0.9-1.0).
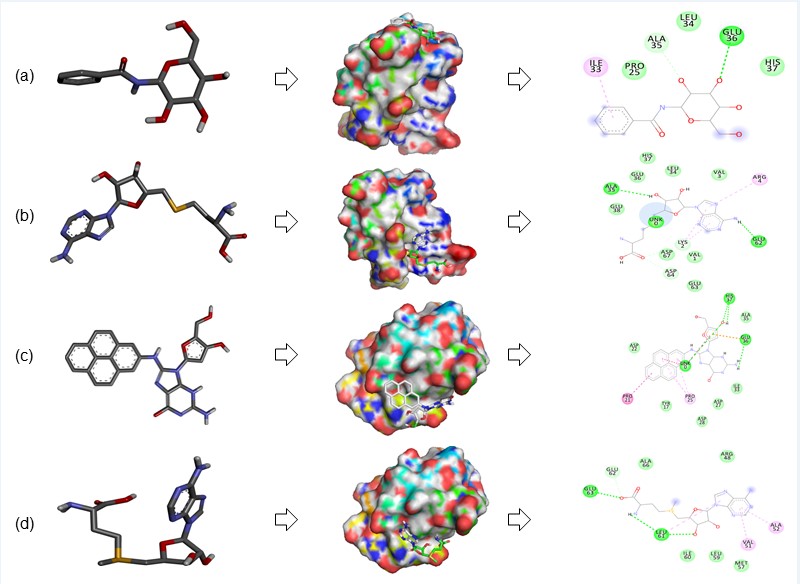
Figure 7: Molecular docking: In this figure, subfigures (a-d) showing the Rv1786 interacting with ligand (a) Rv1786 interacting with 3g2n and 2D diagram analyse the interacting residues. (b) Rv1786 interacting with AdoHcy and 2D diagram analyse the interacting residues. (c) Rv1786 interacting with DGAP and 2D diagram analyse the interacting residues. (d) Rv1786 interacting with S-adenosylmethionine and 2D diagram analyse the interacting residues.
|
S.No. |
Predicted Functional partners |
Information |
PubChem CID |
Molecular Formula |
Molecular Weight |
Score |
|
1 |
S-adenosylmethionine |
Physiologic methyl radical donor involved in enzymatic transmethylation reactions and present in all living organisms. |
1079 |
C15H23N6O5S+ |
399.446 g/mol |
0.795 |
|
2 |
AdoHcy |
Formed from S-adenosylmethionine after transmethylation reactions. |
193 |
C14H20N6O5S |
384.411 g/mol |
0.777 |
|
3 |
Cobalt |
Cyanocobalamin’s structure is based on a corrin ring, which, although similar to the porphyrin ring found in heme, chlorophyll, and cytochrome, has two of the pyrrole rings directly bonded. |
2891 |
C63H89CoN14O14P |
1356.396 g/mol |
0.722 |
|
4 |
Co(NH3)6 |
Causes condensation of DNA in aqueous solution |
122251 |
Co H12N6-4 |
155.071 g/mol |
0.690 |
|
5 |
3g2n |
- |
11243053 |
C13H17NO6 |
283.28 g/mol |
0.444 |
|
6 |
D-GAP |
- |
126100 |
C26 H22N6O4 |
482.5 g/mol |
0.412 |
Table 1: Protein-compound interaction studies: This table enlists the interacting functional partner of Rv1786 protein of M. tuberculosis interconnected with 6 compounds and also shows the score.
|
S.No. |
Model |
RAMPAGE |
Verify 3D |
ERRAT |
|
1 |
QUARK |
Residues in most favoured region 82.5% |
- |
- |
|
Residues in additional allowed region 15.8% |
- |
87.7458 |
||
|
Residues in generously allowed region 1.8% |
86.57 |
- |
||
|
Residues in disallowed region 0.0% |
(PASS) |
- |
||
|
2 |
RaptorX |
Residues in most favoured region 93.0% |
- |
- |
|
Residues in additional allowed region 5.3% |
- |
61.0169 |
||
|
Residues in generously allowed region 0.0% |
23.88% |
- |
||
|
Residues in disallowed region 1.8% |
(FAIL) |
- |
||
|
Residues in most favoured region 75.4% |
- |
- |
||
|
3 |
PHYRE2 |
Residues in additional allowed region 21.1% |
- |
91.5259 |
|
Residues in generously allowed region 3.5% |
62.69% |
- |
||
|
Residues in disallowed region 0.0% |
(FAIL) |
- |
Table 2: Model evaluation by SAVES (Statistical Analysis and Verification Server).
|
S.No |
Drug/Compound |
Docking score |
Interacting bonds |
Interacting amino acids |
|
1 |
3g 2n |
-4.6
|
H bond |
GLU36 |
|
C-H bond |
ALA35 |
|||
|
Pi-Alkyl |
ILE33 |
|||
|
2 |
AdoH cy
|
-5.5 |
H bond |
LEU5 ALA35 GLU36 HIS37 VAL39 |
|
Pi-Alkyl |
LEU34 |
|||
|
3
|
D-GAP
|
-6.3 |
H bond |
ASP49 |
|
Pi-Alkyl |
ARG48 VAL51 ALA52 ILE60 |
|||
|
4 |
S Adenosyl M |
-4.8
|
H bond |
LEU61 GLU63 |
|
C-H bond |
GLU62 |
|||
|
Pi-Alkyl |
VAL51 ALA52 |
Table 3: Docking score of the Rv1786 with interacting amino acids.
Citation: Meena S, Agarwal P, Mishra M, and Meena LS (2019) Prescience of Rv1786 of Mycobacterium tuberculosis H37Rv to assist the pathogenic potentials and significance by computational annotations. Curr Trends Biotechnol Biochem: CTBB-100006
